
Case
125 Diagnosis |
Versión
en Español |
|||
Go back to clinical information and images
Diagnosis: Fabry Disease
Fabry disease is an X-linked recessive lysosomal storage disease that is caused by deficient activity of the lysosomal enzyme α-galactosidase A (α-Gal A), an enzyme that cleaves terminal α-galactosyl residues. This deficiency results in progressive lysosomal accumulation of glycosphingolipid with terminal α-galactosyl residues, particularly globotriaosylceramide (Gb3). Gb3 accumulates in many cells, particularly in renal epithelial cells, endothelial cells, pericytes, vascular smooth muscle cells, cardiomyocytes, and neurons of the autonomic nervous systemlevel (Alroy J, et al. Renal pathology in Fabry disease. J Am Soc Nephrol. 2002 Jun;13 Suppl 2:S134-8. [PubMed link]).
The characteristic appearence of podocytes on light microscopy suggest the diagnosis: podocytes distended with foamy appearing vacuoles. Ultraestructure is useful for diagnosis (Figura 8). Molecular studies are important to confirm the enzimatic alteration.
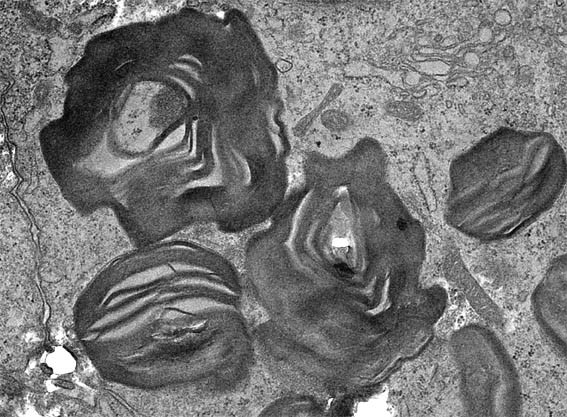
Figure 8. Electron microscopy of a renal biopsy obtained from a 16-yr-old male with Fabry disease. High magnification demonstrates lamellated membrane structures in podocytes, X7,000.
Visit the chapter: Hereditary diseases of our Tutorial (only Spanish version).
Go back to clinical information and images
References
-
Hughes DA. Fabry disease: will markers of early disease enable early treatment and better outcomes? Curr Opin Cardiol. 2016;31(4):434-9. [PubMed link]
-
Biegstraaten M, Arngrímsson R, Barbey F, et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document. Orphanet J Rare Dis. 2015 27;10:36. [PubMed link]
-
Basic-Jukic N, Kes P, Coric M, Basic-Kes V. Renal complications of Fabry disease. Curr Pharm Des. 2013;19(33):6046-50. [PubMed link]
-
Alroy J, Sabnis S, Kopp JB. Renal pathology in Fabry disease. J Am Soc Nephrol. 2002;13 Suppl 2:S134-8. [PubMed link]