Homepage - - - Tutorial Index
Tubulointerstitial diseases
Background, classification, acute pyelonephritis, chronic pyelonephritis, xanthogranulomatous pyelonephritis, malakoplakia, interstitial nephritis associated to drugs, papillary necrosis, interstitial nephritis due to Aristolochia (and Balkan endemic nephropathy), immune mediated interstitial nephritis, IgG4-related kidney disease (tubulointerstitial), acute tubular necrosis, hydropic change, hyaline change, fatty change, hypokalemic nephropathy, Nephrocalcinosis
The tubulointerstitial compartment is affected in all the forms of renal disease. According to the time of evolution and the severity of the lesions, in glomerular and vascular diseases we can find tubular damage, tubular atrophy, edema, interstitial inflammation or fibrosis. Acute interstitial or tubular damage can produce acute renal failure, and chronic changes are a good indicator of irreversible lesions and then they are good prognosis factors in glomerular and/or vascular diseases.
In this chapter we review renal alterations in which tubules and/or interstitium are the primary target of the pathogenic process. The etiology is variable and includes obstructive processes, infections, toxics (including drugs), ischemia, metabolic diseases (like diabetes, hyperuricemia), radiation, damage mediated by immune mechanisms, and infiltration in lymphoproliferative diseases.
Although many of the interstitial inflammatory diseases affect the tubules, a notorious predominance of the interstitial suggests interstitial nephritis. If there are prominent acute tubular changes without interstitial inflammation, acute tubular necrosis is the more probable diagnosis. Due to the frequent involvement of both structures, in many cases is preferable to call the changes as tubulointerstitial nephritis. In chronic changes we find tubular atrophy and interstitial fibrosis doing almost impossible to know the initial target of the aggression.
There is a WHO classification for tubulointerstitial diseases that considers etiology, histology, and clinical features (Fogo AB y Kashgarian M: Diagnostic Atlas of Renal Pathology. Elsevier, Philadelphia, 2005; p. 347):
| WHO
classification of tubulointerstitial diseases |
| Infection: - Acute infectious tubulointerstitial nephritis (TIN) - TIN associated with systemic infection - Chronic infectious TIN (chronic pyelonephritis) - Specific renal infections |
| Drug-induced TIN: - Acute drug-induced tubulotoxic injury - Drug-induced hypersensitivity TIN - Chronic drug-induced TIN |
| TIN associated with immune disorders - Induced by antibodies reacting with tubular antigens - Induced by autologous or exogenous immune complexes - Induced by, or associated with, cell-mediated hypersensitivity - Induced by immediate hypersensitivity (IgE) |
| Obstructive uropathy |
| Reflux nephropathy |
| TIN associated with papillary necrosis |
| Heavy metal-induced tubular and tubulointerstitial lesions |
| Acute tubular injury/necrosis (toxic or ischemic) |
| Tubular and tubulointerstitial nephropathy caused by metabolic disorders |
| Hereditary renal tubulointerstitial disorders |
| TIN associated with neoplastic disorders |
| Tubulointerstitial lesions in glomerular and vascular diseases |
| Miscellaneous disorders - Balkan endemic nephropathy - Toxicity by Aristolochia |
Acute tubulointerstitial nephritis (TIN) is also called pyelonephritis when there is involvement of the collector system and parenchyma by the inflammatory process. It is almost always due to bacterial infection. In 5% of cases the infection involves the kidney by hematogenous dissemination from respiratory tract, skin, or other sites, being some of the more frequent pathogens: Stafilococus aureus, Pseudomonas and Salmonella. In immunocompromised patient Aspergillus or other fungi can be found.
Incidence of pyelonephritis is higher in women, but there is variation with the age: in newborns there is a slightly greater frequency in men due to congenital malformations, and in older than 50 years the frequency newly increases in men due to prostate enlargement. The ascending infections usually are associated to obstructive alterations of the urinary tract, reflux, urinary tract instrumentation, diabetes, pregnancy, and so on. The most common microorganisms come from gastrointestinal tract: E. coli, S. saprophyticus, Klebsiella, Proteus, Serratia and Pseudomonas.
The classic symptoms are fever, lumbar pain and dysuria. In urine culture more than 100,000 colony-forming units are detected in > 80% of patients.
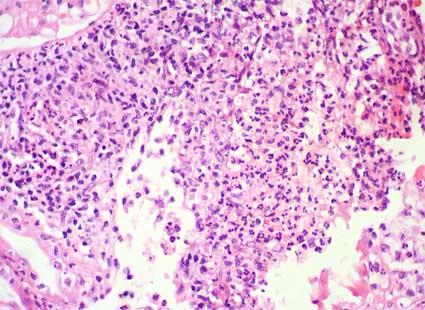
When infection is originated in low urinary tract, polymorphous accumulate in the lumina of tubules, invade epithelium, and migrate to the interstitium. Glomeruli and vessels are usually spared. Tubules can be seen dilated. Zones with inflammatory infiltrates alternate with spared areas. Evident abscesses and necrosis can be seen with a variable frequency (Figure 1). Hematogenous dissemination of an infection to the kidney produces small cortical abscesses without medullary involvement, in opposition to ascending extension of an infection; these abscesses of hematogenous origin are usually glomerulocentric, with minimum tubular involvement (Figure 2). In the inflammatory infiltrates a variable number of lymphocytes are also detected. Plasma cells, macrophages and, sometimes, eosinophils can be prominent. Occasionally eosinophils are so prominent that we can think in allergic or drug-associated tubulointerstitial nephritis.
Figure 1. Acute interstitial Nephritis: there are dense accumulations of polymorphous that destroy tubules and expand the interstitium forming a true microabscess. This case is from a patient with reflux, urinary tract infection, and general symptoms. Due to pyelocaliceal involvement this alteration is also called: acute pyelonephritis. (H&E, X400).

Figure 2. Acute interstitial nephritis originated by hematogenous route usually produces cortical involvement with microabscess, but tubules are usually spared. The acute inflammatory infiltrated tends to be glomerulocentric. Kidney transplanted patient with graft dysfunction, fever, and bacteremia detected in two of three cultures. The renal biopsy only demonstrated cortical acute interstitial inflammation with abundant neutrophils. Renal function was normal after 10 days of antibiotic treatment. (H&E, X400).
In viral infections there are mononuclear inflammatory infiltrates; if the infection is produced by Hantavirus or adenovirus parenchymal hemorrhage can be evidenced. In infections by citomegalovirus and polyomaviruses the characteristic inclusions will be seen (to see infection by polyomavirus and cytomegalovirus in the section of pathology of the renal transplant – Spanish version).
Inmunofluorescencia and electron microscopy
do not show specific findings and usually they are not very useful for the diagnosis.
With some special stains for bacteria, the microorganism can be seen, but, in
the histologic study it is not easy to detect them and to less even classify
them.
In some cases, the infection is located in a segment or lobe of the kidney, forming a well-defined lesion, sometimes with mass characteristics, these cases are known as lobar nephronia. Acute lobar nephronia (ALN), also termed as acute focal nephritis or acute focal bacterial nephritis, is a non-liquefactive localized severe infection of the interstitium of the parenchyma of the kidney involving one or more of the renal lobes (Masood Y, et al. Acute Lobar Nephronia in an infant presented as a renal tumor. Urol Case Rep. 2020;34:101450. [PubMed link]). It is considered a midpoint in the spectrum of upper urinary tract infection between acute pyelonephritis and intrarenal abscess (Ruhela A, Madison G. Acute lobar nephronia: a condition more common in children, a road less travelled in the adults. MOJ Clin Med Case Rep. 2018;8:77-80. [Full text link]).
See Case 193 of our Case Series: Lobar Nephronia.
The term chronic pyelonephritis is used, in general, to designate chronic alterations produced by parenchymal infections (chronic infectious TIN), nevertheless, some authors have used the term in one more indiscriminate way, designating with it some chronic TIN with noninfectious causes. Here, like in most recent texts, we will use this term only for the chronic infectious TIN.
It has been divided, in agreement with physiopathogenic mechanisms, in two types: obstructive (due to urinary tract obstruction) and nonobstructive: reflux nephropathy.
Chronic pyelonephritis can affect people of any age and is more common in women. In many patients there is history of recurrent urinary tract infections, nevertheless, in other it is not so. Sometimes the disease is manifested by renal function alterations without previous history of urinary tract infections. In some patients there is hypertension, and this finding can be related to important renal damage and worse prognosis. In the urinalysis we can detect proteinuria and, sometimes, leukocytes. There is no a specific treatment and the efforts must go to their prevention when there are causes of recurrent infections.
The kidneys have an irregular surface, with depressions and granular aspect (figure 3); cortex can be very similar in aspect to the medulla. In the nonobstructive pyelonephritis parenchymal involvement can be focal, leaving ample areas of spared parenchyma. The collecting system usually is dilated and the parenchyma is narrowed in both types of pyelonephritis, but this dilatation and narrowing are more prominent in obstructive causes, sometimes giving a multicystic appearance. It is very important to determine, macroscopically, that these cavities are interconnected each to other and with the renal pelvis (hydronephrosis) to differentiate it from an actual multicystic kidney disease. When the narrowing and loss of the parenchyma are prominent we say that there is renal atrophy.
Microscopically it is characterized by interstitial fibrosis with inflammatory infiltrates: lymphocytes and plasma cells, and tubular atrophy (Figure 4). Similar changes can be caused by hypertensive nephropathy, chronic glomerulopathies, diabetic nephropathy, and many other alterations. Therefore, to diagnose chronic pyelonephritis we must study very well all the renal compartments, evaluate medullary papillas, pelvis and calyces, and correlate with the clinical and radiological data.

Figure 3. Chronic pyelonephritis, macroscopic aspect of the renal surface of a decapsulated kidney; see the nodularity and the depressed zones that correspond to ample scars.

Figure 4. Chronic pyelonephritis in a patient with congenital vesico-ureteral reflux (nephrectomy). Notice interstitial and periglomerular fibrous areas accompanied of mononuclear inflammatory cells. There are areas without atrophy. In advanced states the histologic aspect is similar to obstructive chronic pyelonephritis. (H&E, X400).
Lesions have a focal distribution. Collapsed tubules mixed with expanded tubules with cylinders of colloid give an aspect similar to the follicles in the thyroid gland: tiroidization. These cylinders contain tubular secretion protein: Tamm-Horsfall’s protein. In cases of obstruction or reflux they can have tubular basement rupture and these proteins (casts) can be seen in the interstitium, they are more evident with PAS stain. Sometimes there is accumulation of polymorphous in interstitium and/or tubules. Atrophic tubules present thinning of their basement membrane. The arteries show unspecific chronic changes: medial thickening and intimal fibrosis.
Glomeruli usually are affected only in advanced stages, when tubular atrophy is prominent. Some glomeruli can be seen normal, others globally sclerosed, some with a retracted tuft, and it is common to see the Bowman’s capsule thickened and periglomerular fibrosis. In reflux nephropathy is frequent to find glomeruli with segmental sclerosis, sometimes in such extension that can make think in a focal and segmental glomeruloesclerosis (as a primary glomerulopathy).
There are no characteristic findings in the immunofluorescence or in electronic microscopy. The presence of significant glomerular deposits of Igs or complement suggests a primary glomerulonephritis.
XANTHOGRANULOMATOUS PYELONEPHRITIS
EIt is a type of chronic pyelonephritis characterized by prominent granulomatous inflammation with abundant lipid loaded macrophages. It is very frequently associated to obstruction and stones. The etiology is not completely understood, but bacterial infections, mainly E. coli, and urinary obstruction are very important in the pathogenesis. Other germs related to the disease have been Proteus, Klebsiella, Pseudomonas and Stafilococus aureus.
The most frequent symptoms are pain, malaise, loss of weight and fever. Most of patients have a history of pyelocaliceal calculi, diabetes or obstruction of the urinary tract. The disease is usually unilateral and it is not associated with renal failure. In some cases we can feel a mass or it can be identified by image studies. Macroscopically the lesion can simulate a carcinoma (and, in some cases, microscopically).
The kidney is increased of size and adhered to the perirenal fat, with thickened capsule; pyelocaliceal cavities appear dilated and usually occupied by purulent material and one or several calculi. Yellow, soft and friable areas are evidenced. There can be abscesses in the renal parenchyma. Some times the lesion is focal. [See several macroscopic images (Link)]
Microscopically the yellow areas are formed by pile of large lipid loaded macrophages: xanthoma cells (foam cells), mixed with smaller macrophages, lymphocytes, plasma cells, and polymorphous. Multinucleated giant cells can be seen. Areas with necrosis and calcifications are also seen. In other parenchymal areas there is inflammation, interstitial fibrosis and tubular atrophy. The renal capsule appears fibrotic and adhered to perirenal fat.
This disease is more frequently found in the bladder and gastrointestinal tract, but some times it is diagnosed in renal pelvis or, more rarely, renal parenchyma. Malakoplakia in kidney and urinary tract affects women more frequently, and age of diagnosis is often in the fifth decade of the life. It is unilateral in most of cases and it is not associated with calculi, but with urinary tract infections.
It is considered that the first human case of malacoplakia was seen by the German professor D. von Hansemann (1901). Professor von Hanseman then spoke to his assistant Dr Gutmann and provided him with details of his case, as Dr Michaelis, an expert biochemist, had agreed to study the disease further in a collaborative venture. Not knowing how history would be written, von Hansemann published his paper in 1903: von Hansemann D. Über Malakoplakie der Harnblase. Virch Arch Path Anat 1903; 173: 302–8, a year after Michaelis and Gutmann described the first human case: Michaelis L, Gutmann C. Ueber Einschlüsse in Blasentumoren. Ztschr Klin Med 1902; 47: 208–15. (Dasgupta P, Womack C, Turner AG, Blackford HN. Malacoplakia: von Hansemann's disease. BJU Int. 1999;84(4):464-9. [PubMed link])
The term malakoplakia (some authors write "malacoplakia") was coined by von Hansemann in 1903 and is derived from the Greek words malakos (soft) and plakos (plaque).
Malakoplakia is the result of an acquired defect in macrophage function causing impairment of bactericidal activity. The precise mechanism is unknown. The large macrophages that are present at sites of infection (von Hansemann cells) exhibit numerous secondary lysosomes containing partially digested organisms. Fusion and calcification of these lysosomes results in the formation of intracytoplasmic bodies called Michaelis-Gutmann bodies, considered pathognomonic of malakoplakia. Approximately 50% of patients present alterations of the immune system: hypogammaglobulinemia, immunosuppressor therapy, rheumatoid arthritis, AIDS, or others.
The most frequent symptoms are fever and pain in the flank. There can be pyuria, proteinuria and/or hematuria. Like in xanthogranulomatous pyelonephritis, a mass can be identified and it can simulate a neoplasm.
In the kidney there can be extensive or focal involvement, with yellow or brown areas under the pyelocaliceal epithelium or under renal capsule. Deep parenchymal lesions appear, at cut, as areas with renal distortion.
Microscopically the lesions are constituted by foam cells (macrophages) with eosinophilic granular cytoplasm (the von Hansemann cells) (Figure 5). In many of this cells there is cytoplasmic basophilic 1 – 10 microns inclusions; these inclusions are intensely PAS positive, hematoxylinic, and positive with the von Kossa stain (for calcium) (Michaelis-Gutmann bodies) (Figure 6). Occasionally these bodies can be found in the interstitium. Michaelis-Gutmann bodies can appear laminated, homogeneous, or with a dense central core with a targetoid appearance. In other areas of the renal parenchyma there are unspecific changes. [Several images of malakoplakia in bladder, colon and lung (link)] [Image of malakoplakia in prostate (link)]

Figure 5. Pile of large, eosinophilic, granular cells: von Hansemann cells. In the cytoplasm of these histiocytes there are basophilic inclusions with a variable size: Michaelis-Gutmann bodies, pathognomonic of the disease. (H&E, left, X400; right, X1.000)

Figura 6. Histiocytes with many Michaelis-Gutmann bodies; these appear basophilic and size varies between 1 micron and 10 microns. In many there is a central core and a targetoid appearance (green arrows), in others appearance is homogeneous (blu arrows). In some cells there are several inclusions (red arrow), and ocasionally this bodies can be seen extracellular. (H&E, both X400).
By electron microscopy, the inclusions have crystalline structure, with a central dense body surrounded by a clear halo and a laminated peripheral ring. In the center of the inclusion there can be material that is similar to bacterial fragments (Kern WF, Silva GF, et al, Atlas of Renal Pathology. W. B. Saunders Company, Philadelphia, 1999, p. 147).
There is a disease denominated megalocytic interstitial nephritis, characterized clinically and morphologically by alterations similar to those of malakoplakia, but in which Michaelis-Gutmann bodies are not identified. It is not known if it is a diverse disease or is a morphologic variant of malakoplakia.
[A
revision of malakoplakia and images in eMedicine©
(link)]
[See Case 35 of our Case Series] [See Case 106 of our Case Series] [See Case 129 of our Case Series]
ACUTE INTERSTITIAL NEPHRITIS ASSOCIATED TO DRUGS
Drugs are the main cause of interstitial nephritis (IN). The list of drugs causing IN grows constantly. The most common include: antibiotics (penicillins, cephalosporins, sulfonamides, tetracyclines, vancomycin, ciprofloxacin…); non-steroidal anti-inflammatory (NSAID) (salicylic acid, ibuprofen, naproxen, indomethacin, sulindac, mefenamic acid…); diuretics (thiazides, furosemide, triamterene, chlortalidone); and others (acetaminophen, captopril, cimetidine, ranitidine, phenobarbital, phenytoin, lithium, interferon, acyclovir, cyclosporine…).
There are several mechanisms related to drugs associated IN. The inhibition of synthesis of prostaglandins by NSAID can alters the distribution of the renal sanguineous flow; many cases are related to allergic reactions; in other cases deposits of immunoglobulins in the tubule basement membranes (TBM) are demonstrated, which could be due to union of the drug with the TBM and later fixation of an antibody against this medicine.
The main clinical manifestation of the disease is renal failure. In acute IN the starting is abrupt and usually we can detect one or several drugs used before the episode. In cases of chronic IN the clinical presentation is gradual. In some cases there is fever and malaise; in other pain (perhaps by distension of the renal capsule as a result of inflammation and edema). In some cases of acute IN there is oliguria. Glucosuria, aminoaciduria, phosphaturia, renal acidosis, and loss of sodium or potassium can be detected, as a result of the tubular injury. There is proteinuria, sometimes in nephrotic range. In hypersensitivity reactions very few patients show systemic symptoms like fever and cutaneous alterations; around half of the patients will present some degree of eosinophilia; sometimes eosinophils are detected in the urine, but it is not specific of the hypersensitivity reactions.
Microscopically the interstitium appears enlarged by edema (acute IN), mononuclear inflammatory infiltrates, or fibrosis (in chronic IN). The feature of chronic IN is fibrosis. The interstitium with edema is seen more pale and with fibrillary appearance. Fibrosis is more solid and more intensively stained with H&E or trichrome (blue or green). The inflammatory cells are predominantly plasma cells, lymphocytes, and, in some cases, eosinophils. There can be also macrophages, neutrophils, and multinucleated giant cells; in some cases there are non-caseous granulomas. The infiltrate is in many cases characteristically focal and more prominent in the corticomedullary union, often surrounding individual tubules. We can see pile of eosinophils, but if there are microabscesses of neutrophils we must think in an infectious cause.
Tubules present very variable lesions that can be focal. Sometimes there is frank tubular necrosis, but in others cases attenuation or loss of the brush border is the only change seen (better evidenced with PAS stain). The tubular epithelial cells can be seen free in the lumen of the tubule or forming cellular casts. We will also find, in some cases, regenerative changes: mitosis, enlarged nuclei with prominent nucleoli, and binucleation. We can see infiltration of lymphocytes or neutrophils in the tubular epithelium: tubulitis. We can also identify rupture and laminated aspect of the TBM.
Glomeruli and vessels typically appear normal in acute IN. In chronic IN there will be glomerulosclerosis and angiosclerosis in a variable extension according to the tubulointersticial damage.
By immunofluorescence usually there are no immune deposits; fibrin or fibrinogen is detected in the interstitium. In some few cases linear staining (sometimes granular) for IgG and C3 is detected in TBM, mainly in IN associated to methicillin, penicillins and cephalosporins. By electron microscopy there are not specific findings; in some cases there are electron-dense deposits in the TBM .
See Case 59 and Case 68 of our Case series.
________________________________________________________________
A syndrome of IN associated with uveitis has been described: Tubulointerstitial nephritis and uveitis syndrome (TINU). It happens in children and adults and it is commonest in women. The cause is unknown but it is related to an immunological mechanism. The histologic picture shows interstitial inflammation by lymphocytes, plasma cells, monocytes, neutrophils, and sparse eosinophils. They can have granulomas. There is tubular necrosis and epithelial regeneration. There are not glomerular or vascular lesions. Immunofluorescence is negative. Treatment is steroids and the prognosis is favorable (Goda C, et al, Am J Ophthalmol 200;140:637 PubMed link; Sessa A, et al, J Nephrol 2000;13:377 PubMed link; Hinkle DM, Foster CS. Tubulointerstitial nephritis and uveitis syndrome. Int Ophthalmol Clin. 2008;48(2):9-13. [PubMed link]).
Another described syndrome is IN and uveitis associated with granulomas in the bone marrow, hypergammaglobulinemia and increased erythrosedimentation, is known as syndrome of Dobrin (Kern WF, Silva GF, et al, Atlas of Renal Pathology. W. B. Saunders Company, Philadelphia, 1999, p. 139).
See a very interesting case with its discussion: Neilson EG, Farris AB. Case 21-2009 — A 61-Year-Old Woman with Abdominal Pain, Weight Loss, and Renal Failure. N Eng J Med 2009;361:179-187. [Link to the case in NEJM].
________________________________________________________________
Papillary necrosis occurs by three main causes: 1.) Use of analgesic by long time; 2.) acute pyelonephritis, especially in diabetics or patients with urinary tract obstruction; and 3.) in sickle cell anemia. Other causes can be infections, renovascular disease, and graft rejection. Ischemia seems to be a key factor in its development. The analgesics more related to papillary necrosis have been phenacetin, acetaminophen, acetyl salicylic acid, caffeine and codeine. The disease usually occurs in the event of prolonged and combined use in high doses. Analgesic nephropathy has been associated, in addition, with urotelial carcinoma, especially in smokers. The clinical presentation is highly variable, but usually there is renal failure with insidious onset and progression.
Papillary necrosis can be uni-or bilateral. Macroscopically the kidneys show yellow and friable papillae surrounded by a hyperemic edge. Necrotic papillae can be seen firm, sclerosed and retracted; or they can fall in pelvis and show a concave bed. In analgesic nephropathy the involvement is bilateral and in all the papillae, although they appear in different stages of disease evolution. In cases of high phenacetin use a yellow or brown color in the mucosa of pelvis of pyramids can be seen.
Microscopically there is ischemic necrosis and neutrophils surrounding these zones. In analgesic nephropathy there is thickening of capillary basement membranes in peritubular and urinary mucosa capillary, and basement membrane of the ascendant portion of the limbs of Henle. There is greater involvement in the deep part of the medulla; the peripheral parts of the pyramids, that receive sanguineous flow of the columns of Bertin, appear more preserved. Areas with necrosis and tissue loss are covered by collecting ducts epithelium.
There are not specific features by inmunofluorescencia or electron microscopy .
_________________________________________________________
Occasionally there is renal involvement by sarcoidosis that must be differentiated from granulomatous infections like tuberculosis or mycosis. Wegener’s granulomatosis not very often gives granulomas in the kidney.
_________________________________________________________
INTERSTITIAL NEPHRITIS BY ARISTOLOCHIA (AND BALKAN ENDEMIC NEPHROPATHY))
From the Fifties an endemic nephropathy has been described in Croatia, Bosnia, Serbia, Bulgaria and Romania: Balkan endemic nephropathy. For many years speculated about a possible association with chronic poisoning by heavy metals, silica, toxins of fungi and other toxins of plants, virus and deficiencies in the diet. At the moment, many works have demonstrated the association with the plant Aristolochia (in the Balkan specifically the species clematitis). Hranjec et al. have hypothesized that “flour used to bake bread, a dietary staple in the endemic region of Croatia, is derived from wheat grain which, in the past, is likely to have been contaminated with seeds of A. clematitis during harvesting” (Hranjec T, et al, Croat Med J. 2005;46:699 PubMed link / Free full text).
A good review: Bamias G, Boletis J. Balkan Nephropathy: Evolution of Our Knowledge. Am J Kidney Dis 2008;52(3):606-616 [Free full text].
Other species of Aristolochia have been associated with toxic nephropathy in Europe (extensively studied in Belgium) and Asia. It is possible that this one, or other plants, produce similar alterations in the American continent. This disease, or similar, has been called “Chinese-herb nephropathy” (de Jonge H, Vanrenterghem Y. Aristolochic acid: the common culprit of Chinese herbs nephropathy and Balkan endemic nephropathy. Nephrol Dial Transplant. 2008;23:39-41. [PubMed link]; Cosyns JP. Aristolochic acid and 'Chinese herbs nephropathy': a review of the evidence to date. Drug Saf. 2003;26:33-48. [PubMed link]; Debelle FD, Vanherweghem JL, Nortier JL. Aristolochic acid nephropathy: A worldwide problem. Kidney Int. 2008 74:158-159 [PubMed link])
La The clinical presentation is related mainly to slowly progressive chronic renal failure. In some cases the patient is relatively healthy, but begins to feel bad and when consulting to its doctor chronic advanced renal failure is documented. There are unspecific general symptoms.
The nephropathy described in these patients is predominantly interstitial. There is extensive interstitial fibrosis that, at least initially, is associated with mononuclear inflammatory infiltrates. When advancing the process diminishes the inflammatory infiltrate and the fibrosis becomes diffuse, with severe tubular atrophy. In these biopsies is striking the good preservation of the glomerular structure until advanced states of the process .
TUBULOINTERSTITIAL NEPHRITIS MEDIATED BY IMMUNE MECHANISMS
These cases of tubulointerstitial nephritis (TIN) can be secondary to anti-GBM disease, lupus nephritis, Sjögren’s syndrome, and other diseases with formation of immune complexes. The histologic changes are unspecific and usually are accompanied by interstitial or tubular basement membrane immune deposits.
TIN with anti-tubular basement membrane (anti-TBM) antibody disease: It is rare and presents with acute or chronic kidney injury. Patients may be of any age and usually have polyuria and polydipsia. Microhematuria and proteinuria (sometimes nephrotic-range) are also seen. Light microscopy: There is an interstitial mononuclear infiltrate with lymphocytes, plasma cells, and macrophages, with occasional neutrophils. Variable interstitial eosinophils can also be present with tubulitis with associated acute tubular injury and interstitial edema, rarely with interstitial giant cells. Glomeruli and arteries show nonspecific changes. The extent of interstitial fibrosis and tubular atrophy varies depending on the chronicity of anti-TBM disease at the time of biopsy. Immunofluorescence microscopy: There is strong diffuse linear staining of TBMs for IgG, with variable C3 staining. Electron microscopy: No deposits are seen. Etiology/Pathogenesis: Primary anti-TBM disease is related to an autoantibody to a protein called tubulointerstitial nephritis antigen, which is only expressed in the kidney. This antigen is a 58-kDa noncollagenous protein found in the proximal TBM. It is a regulatory protein in tubulogenesis that interacts with type IV collagen, laminin, and integrins. In some patients, drug exposure may have triggered autoantibody formation. Serum anti-TBM antibodies are detected [Lusco MA, et al. AJKD Atlas of Renal Pathology: Anti-Tubular Basement Membrane Antibody Disease. Am J Kidney Dis. 2017 Jul;70(1):e3-e4. [PubMed link] [Full text link]].
It also has been described a TIN mediated by T cells, many of these cases associated with uveitis (TINU syndrome, see above).
IgG4-RELATED KIDNEY DISEASE (TUBULOINTERSTITIAL)
Immunoglobulin G4-related diseases (IgG4-RDs) is a clinical entity that often involves multiple organs and is characterized by high levels of serum IgG4, dense infiltration of IgG4+ cells and storiform fibrosis. Cellular immunity, particularly T-cell mediated immunity, has been implicated in the pathogenesis of IgG4-RDs. The most frequent renal manifestations of IgG4-RD are IgG4-related tubulointerstitial nephritis, membranous glomerulopathy and obstructive nephropathy secondary to urinary tract obstruction due to IgG4-related retroperitoneal fibrosis. IgG4-RD diagnosis should be based on specific histopathological findings, confirmed by tissue immunostaining, typical radiological findings and an appropriate clinical context. The first line treatment is the steroids with two warnings: Steroid resistance and relapse after discontinuation. In the case of steroid resistance, B cell depleting agents as rituximab represent the second-line treatment. In the case of relapse after discontinuation, steroid treatment may be associated with steroid sparing agents (Salvadori M, Tsalouchos A. Immunoglobulin G4-related kidney diseases: An updated review. World J Nephrol. 2018;7(1):29-40. [Full text link];Capecchi R, et al. Renal Involvement in IgG4-Related Disease: From Sunlight to Twilight. Front Med (Lausanne). 2021 Mar 31;8:635706 [PubMed link]).
See Case 185 of our Tutorial: Tubulointerstitial IgG4-related Kidney disease.
LAcute tubular necrosis (ATN) produces acute renal failure. In cases of rapid loss of function is usual to take a renal biopsy, reason why we find biopsies with this diagnosis with relative frequency. ATN occurs by two main causes: ischemia and toxicity. In many cases there is not a true necrosis of the tubules, but alterations of the epithelial function with the consequent hydroelectrolytic changes and reaction of the epithelium trying to repair the injury.
Ischemic acute tubular necrosis
It is one of the most frequent causes of acute renal insufficiency. It can be
seen in a great variety of conditions that diminish the renal perfusion, many
associated with hypovolemia. Some of the most frequent causes of ischemic ATN
are: surgical complications, trauma or extensive burns, pancreatitis, hemolytic
reactions to transfusions, sepsis, obstetrical complications, and rhabdomyolysis.
The histology is similar in anyone of these causes.
In many of the cases there is oliguria, but in others there is non-oliguric renal failure. There is loss of the capacity to urine concentration and there is high concentration of urinary sodium. In many of the cases there is a precipitating factor one or several days previous to renal failure. There is increase of the BUN and creatinine, proteinuria (usually nonselective and mild), granular and hyaline cast, non-concentrated urine, and a high sodium fraction excretion rate; usually there is not hematuria.
The kidneys are increased in size and pale, there is widening of the cortex, the medulla appear congested, and there is increase in the corticomedullary differentiation.
Tubules: Histologic changes of tubular epithelium are usually more subtle in the ischemic type that in the toxic injury. In addition, the sites of tubular damage along the nephron differ between the two forms. In the ischemic lesion, tubular damage is patchy, affecting relatively short lengths of the straight segments of the proximal tubule and focal areas of the ascending limbs of the loops of Henle. In the toxic form, the tubular epithelial damage is more extensive along segments of the proximal tubule.
There is loss of some cells with shedding of both necrotic and viable epithelial cells into the tubular lumen, leaving tubular basement membranes denudated. Some tubules appear dilated and with flattened epithelium (Figures 7, 8 and 9). In proximal tubules the brush border is often thinned or absent (better seen with PAS stain), adopting a pattern similar to distal tubules: “distalization”. There is formation of small bleb in the luminal edge of the cells (“blebbing”) that detach and fall in the lumen (Figure 10). Tubular damage is patchy and the segments more injured are in the third portion of the proximal tubule (S3) and the ascending limb of the loop of Henle. In distal tubules the epithelium is flattened and dilated. There are granular, hyaline, cellular, and/or pigmented (brown tone) casts in distal tubules, mainly in collecting ducts. We can see also leukocytes in tubular lumina. In the interstitium there can be edema, inflammatory infiltrates and even granulomas.

Figure 7. Acute tubular necrosis with dilatation of tubules, epithelial flattening, brush border loss in proximal tubules, and shedding of cells in some of them. (Masson trichrome, X300).

Figure 8. Severe acute tubular necrosis in which masses of cells (“fallen”) are observed in the lumina of tubules; there is complete denudation of some of them and almost complete obstruction of their lumina. This image is from a biopsy of a kidney allograft (one post-transplantation week) from a non-beating heart donor; kidney allografts from this donor type usually present severe ATN, as this case, nevertheless, after their recovery, two or three weeks later, these kidneys usually have an excellent prognosis. (Case of Department of Anatomia Patológica, Hospital Clínico San Carlos, Madrid, Spain, with authorization of Dr. Julia Blanco). (Masson’s trichrome, X400).

Figure 9. Another case with severe ATN in a kidney allograft receptor of a non-beating heart donor. See the amorphous, granular masses, with cellular detritus, that fill some tubules. In spite of the severity of the injury, these patients recover their function without residual chronic damage. These so severe changes of ATN are rare in native kidneys, but it can be also seen in receptors of brain death donors with prolonged periods of cold ischemia or organ preservation problems. (Case of Department of Anatomia Patológica, Hospital Clínico San Carlos, Madrid, Spain, with authorization of Dr. Julia Blanco). (Masson’s trichrome, X400).

Figure 10. Tubules with initial changes of ATN. There are cytoplasm fragment projections towards tubular lumina and loosening of some of these microvesicles (“blebbing”); the brush border is lost and some free cells are in the lumina (arrow). (Masson’s trichrome, X400).

Figure 11. Regenerative changes in a patient with acute tubular necrosis. Notice that most of tubular cells in this microphotography present high nucleus/cytoplasm relation, large nuclei with prominent nucleoli that indicate cellular synthetic activity and mitosis (arrow). These changes indicate the favorable response of the epithelium towards recovery. ATN is a lesion that, if not associated to prolonged injury or tubular basement membrane damage, is completely reversible. (Case of Department of Anatomia Patológica, Hospital Clínico San Carlos, Madrid, Spain, with authorization of Dr. Julia Blanco). (Masson’s trichrome, X400).
In peritubular vessel of the external medulla we can see abundant intracapillary nucleated cells; it is believed that they are hematopoietic precursory cells originating in bony marrow; in initial phases we can see lymphocytes. The mechanism of accumulation of these cells is not very clear but when they are evidenced it is a good histologic key for ATN diagnosis(Jennette JC et al, Heptinstall's Pathology of the Kidney, 5º ed. Lippincott-Raven, Philadelphia, 1998, p. 877).
The initial changes in ischemic ATN are attenuation or loss of the brush border in proximal tubules and, in some cases, cytoplasmic vacuolization or intracellular edema. In cases of hemolysis or extensive muscular damage (as in malignant hyperthermia or electrical burns) pigmented cylinders of hemoglobin or myoglobin are detected (Figure 12). In some túbulos we can see intraluminal calcium grains.

Figure 12. Biopsy of a kidney donor from a patient died of malignant hyperthermia. Tubular lumina are occupied by red grains of myoglobin (arrows). Both kidneys were discarded for transplantation. (Case of Department of Anatomia Patológica, Hospital Clínico San Carlos, Madrid, Spain, with authorization of Dr. Julia Blanco). (Masson’s trichrome, X400).
When regenerative changes of the epithelium begin, the cytoplasm of epithelial cells becomes basophilic, the nuclei become hyperchromatic, with increase of size and presence of nucleoli; there is increase of the relation nucleus/cytoplasm and mitosis (Figure 11).
There are not evident glomerular changes; sometimes they appear without blood in capillary and contracted, with expansion of the urinary space. The Bowman’s capsule epithelium can have a cuboidal aspect: Tubularization of the parietal epithelium.
There are not immunopathological alterations (by immunofluorescence). With electron microscopy changes in the brush border are more evident, there is simplification of the lateral interdigitating projections of lateral borders of tubular cells, and the apical cellular membrane shows small cytoplasmic projections: blebbing (in many cases they are also seen with light microscopy). There are cytoplasmic vacuolization, mitochondrial changes, and inclusions that probably represent lipofuscin. There is apoptosis in some cells. In cases of initial ATN that is difficult to diagnose by light microscopy, the ultrastructure is very useful in the definitive diagnosis.
Toxic acute tubular necrosis
It can be caused by poisoning with heavy metals such as mercury or arsenicals,
or with organic compounds such as carbon tetrachloride. Nevertheless, most of
cases currently seen are associated to aminoglycoside antibiotics, antineoplastic
agents such as cis-platinum, and substances extracted from plants and commercialized
as “natural products” (herbal medications). In toxic tubular damage
the epithelial lesions are histologically more evident than in ischemic lesions
and proximal tubule is more extensively involved. The degree of involvement
of the segments varies with the specific toxin.
The clinical picture is similar to ischemic ATN, with rapid onset renal failure; in cases of industrial exposure the beginning can be slow.
Histologically there is extensive epithelial necrosis that tends to involve all the nephron. Proximal tubule is the more involved with basal denudation and lumen occupied by cellular detritus. Calcification of this intratubular necrotic material can be seen as soon as 1 or 2 days. Regenerative changes are also evident, epithelium is initially flat and progressively it increases his height, with nuclear changes and mitosis; there is heterogeneity in cell size. Some toxins give microscopic alterations different to others, according to their mechanism of injury.
In cases of acute toxicity by lead we can identify dark intranuclear inclusions: dense bodies formed by complexes of lead and other molecules. In nephrotoxicity by glycol, oxalate crystals are observed (an unspecific feature). In nephrotoxicity by aminoglycosides there are lysosomal myeloid bodies (electronic microscopy).
In chronic exposure to lead, mercury, cadmium, platinum, gold, lithium, silver, iron and copper there is a unspecific chronic tubulointerstitial nephritis. There are proximal tubular damage and intranuclear inclusions formed by complexes metal-metalothianides, synthesized by the tubular cells. The mechanism is related to filtration and later reabsorption and accumulation in the epithelial cell.
In indinavir nephropathy there is formation of crystals of this drug that is filtered and excreted in the urine. Dehydration, elevated pH, high levels of the drug, and medicine interactions can diminish their solubility and increase the formation of crystals. Indinavir is an inhibitor of proteases, used in infection by HIV-1. It can produce ATN with crystalluria. The crystals obstruct tubules and generate peritubular inflammatory reaction. The crystals have needle form and they form irregular intratubular piles (Reilly RF, et al, Am J Kidney Dis 2001;38:E23 PubMed link; Herman JS, et al, J Antimicrob Chemother 2001;48:355-60 PubMed link / Free full text).
La ATN, ischemic or toxic, is a completely reversible process; the prognosis depends on involvement of other organs or extrarenal complications. If the tubular injury persists more than a month the possibility of chronic renal damage increases.
In kidney allografts from non-beating heart donors the severe changes are very noticeable and regenerative changes are prominent. Sometimes, the regenerative changes simulate cytopathic viral changes. Even so recovery of tubular epithelium is complete.
OTHER TUBULAR CHANGES
Also called “osmotic nephrosis”. It is observed in patients who have received hypertonic solutions as sucrose, mannitol, or dextran, for treatment of cerebral edema or increased intracranial pressure, or to expand volume. Usually it is not associated with alterations of the renal function. In some cases of patients with cardiac or liver failure and systemic infections, some authors consider that it can be an initial change of ischemic or toxic injury.
Tubules appear pale and tubular cells have a clear cytoplasmic aspect, with ill-defined vacuoles. Brush border is conserved. The changes are more prominent in proximal tubules and, in initial phases, are evidenced in the straight portion (S3). It has been demonstrated, experimentally, that this change results for expansion of lysosomes (Figure 13) (Dickenmann M, Oettl T, Mihatsch MJ. Osmotic Nephrosis: Acute Kidney Injury With Accumulation of Proximal Tubular Lysosomes Due to Administration of Exogenous Solutes. Am J Kidney Dis 2008 Mar;51(3);491-503. [PubMed link])

Figure 13. Proximal tubules with vacuolated cytoplasm, brush border attenuation, and nuclear features preserved, without basal denudation or shedding of cells. This change usually is associated with use of hypertonic solutions in the patient, but also we can see it in initial phases of an acute tubular necrosis. (Masson’s trichrome, X400).
Hyaline change or formation of hyaline droplets
It is the accumulation of eosinophilic, PAS-positive and methenamine-silver-positive droplets that fill and expand the cytoplasm of tubular cells. They vary in size and they are observed more frequently in proximal tubules, although they can be also seen in other portions of tubules and in podocytes. These droplets are due to protein reabsorption. Droplets can be positive, by inmunofluorescencia, for many types of proteins, including albumin, immunoglobulins, complement fractions, and others. Its presence does not indicate alteration of the tubular function and, on the contrary, it is an indicator of normal tubular reabsorption (Figure 14).

Figura 14. Hyaline droplets formation is a very common feature in patients with some degree of proteinuria. In many cases of nephrotic syndrome they are prominent and they can be positive for immunoglobulins on immunofluorescence. These droplets indicate an adequate tubular function. Hyaline droplets are best seen with PAS and silver stains. (Masson’s trichrome, X400).
Normally the kidney contains a 2.3% of its weight as lipids, mainly in the medulla. In patients with proteinuria or nephrotic syndrome, tubular cells reabsorb lipid and it can be accumulated in its cytoplasm giving them a vacuolated aspect. These cells are observed in the urinary sediment as oval fat bodies. When the vacuoles are small the cell has an aspect similar to hydropic change and to differentiate it is necessary carry out a stain for fat detection in frozen tissue.
Hypokalemic nephropathy (or nephrosis)
It is caused by chronic, long-standing hypokalemia, such as in patients with chronic laxative abuse. Large vacuoles appear in the cytoplasm of cells of proximal convoluted tubules. These vacuoles do not contain proteins, lipids or glycogen. The mechanism of their formation is unclear.
View this topic in Discussion of Case 60 of our case series.
Recent bibliography
Tubulointerstitial nephritis
Acute tubular necrosis
Homepage - - - Tutorial Index