
Homepage - - - Tutorial Index
Vascular Diseases
Hypertension, Bartter syndrome, fibromuscular dysplasia of the renal artery, cholesterol embolism, cortical necrosis, hemolytic-uremic syndrome / thrombotic thrombocytopenic purpura, preeclampsia/eclampsia and large - medium vessels vasculitides
Glomerular filtration is dependent on an adequate blood supply to the capillaries of the tuft; therefore, vascular lesions, in addition to tissue damage, alter the corporal volume and electrolyte balance. The kidneys receive 25% of the cardiac output, a larger proportion than any other organ, thus vascular lesions significantly affect these organs. Renal vascular diseases are usually the manifestation of a systemic vascular disease. Here we will focus on renal changes in vascular diseases, and not on extrarenal involvement.
RENAL INVOLVEMENT IN SYSTEMIC ARTERIAL HYPERTENSION
Systemic arterial hypertension (high blood pressure - HBP) has been classified according to etiology in essential, primary or idiopathic (unknown cause) and secondary (caused by other conditions). It has also been classified as mild, moderate and severe (or in gdegrees), according to values of systolic and diastolic pressure. Sometimes the term benign hypertension is used to call the mild to moderate hypertension. Malignant hypertension is defined by rapid damage (or acute) of one or several target organs (with papilledema), rather than an exact number of blood pressure. Initially we will refer to renal changes in chronic hypertension (not malignant) and in a different section, in this page, we will refer to renal changes in malignant hypertension. The renal involvement in HBP is also known as hypertensive nephropathy.
HBP is a risk factor for chronic kidney disease and other organs damage, however, is usually asymptomatic until advanced stages of tissue lesion. The physiopathology of hypertension is complex and widely discussed in other texts. Here we shall only present renal alterations secondary to it.
When the disease has produced significant
renal injury, the organ is reduced in size and weight, there is usually a granular
capsular surface and depressions may be secondary to fibrosis by ischemia: scars;
these changes are very characteristic of hypertensive nephropathy. The cortex
is thin and often there are simple cysts with variable size.
Histopathology
The renal histological changes of chronic hypertension are not specific. Glomerular and tubulointerstitial lesions are of ischemic type and its microscopic appearance is similar in all forms of vascular injury with secondary ischemic renal damage. The glomeruli are retracted, capillary walls are thick and wrinkled with rolling or “accordion-shape” and there is a relative increase in the Bowman’s space. Changes in the capillary walls become more evident with PAS or silver staining (Figure 1). In some cases focal and segmental sclerosing lesions can be evidenced and they can be associated with proteinuria (secondary focal and segmental glomeruloesclerosis). In more advanced stages collagen deposits are evidenced in the inner portion of the Bowman’s capsule (intracapsular fibrosis). Initially, collagen is deposited near to the vascular pole and gradually fills the entire circumference of the Bowman’s space, the tuft is compressed by the fibrous tissue and the glomerulus ends in ischemic type global sclerosis (Figure 2). In cases of global glomerular sclerosis the fibrous tissue and the sclerosed tuft stain with a similar appearance with H&E, but with PAS and methenamine-silver there is a evident contrast between the scarring collagen that occupies the Bowman’s space (interstitial type collagen: fibrosis), which is weakly positive or negative with both stains, and the type of collagen in the compressed glomerular tuft: sclerosis, which is intensively positive with PAS or silver. In many cases, with the methenamine-silver staining there are portions or fragments of glomerular basement membrane (GBM) in the center of the sclerosed glomerulus (Figure 2). Gradually, over time, the residual tuft collagen tends to lose and, finally, there is only fibrous tissue and it may be difficult or impossible to recognize that there was a glomerulus.
Figure 1. Initial glomerular ischemic changes; capillary walls appear retracted and irregulars, there is decrease in the size of the tuft and a wide Bowman’s space; there is not still sclerosis. Theoretically, these changes are reversible. In many cases, we also identify tubular basement membranes with similar appearance (not shown in photo). (Methenamine-Silver, X400).
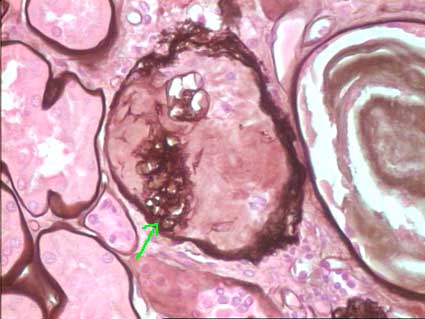
Figure 2. Glomerulus with global sclerosis. Residual fragments of the tuft can be still identified with silver stains (arrow). The material occupying the Bowman’s space is type interstitial collagen, with negative staining. PAS staining gives a similar appearance. (Methenamine-Silver, X400).
There are interstitial fibrosis and tubular atrophy in multiple areas. In preserved tissue tubules usually appear dilated (compensatory dilatation or "super tubules"). In atrophic tubules its lumen is narrowed, the epithelial cells are low and sometimes with clear cytoplasm and the brush border of proximal tubules disappears. The tubular basement membranes may be thickened, as in glomerular capillaries, and they appear irregular, wavy or wrinkled with PAS or silver staining. In the fibrotic interstitium, as in fibrosis of other causes, there may be mononuclear inflammatory infiltrates (Figures 3 and 4).

Figure 3. Kidney of a patient with chronic Hypertension who died due to cerebrovascular disease. There are multiple areas of fibrosis, mild scattered mononuclear inflammatory infiltrate, focal global glomeruloesclerosis (blue arrows) and intimal fibrosis in arteries (green arrows). (Masson's trichrome, X200).

Figure 4. Scared area which takes a linear pattern from the renal capsule (right) which has a depression (arrow). This aspect indicates obstructive lesion in the artery that irrigate the fibrotic area. If in the histologic evaluation of a possible donor the sample is taken from this area, the damage would be overestimated; biopsies from kidney donors should be taken from areas with macroscopically normal appearance. (Masson's trichrome, X200)..
In some cases there is hypertrophy of the yuxtaglomerular apparatus, however, this hypertrophy is difficult to see when histologically mild, being necessary computer image analyses (histomorphometry) to determine it with accuracy. Severe degrees of hypertrophy are easily identified in the histologic evaluation with conventional light microscopy.
Arteries: there is concentric thickening of the intima by deposition of collagen: intimal fibrosis; muscular hyperplasia of the media; the lumens appear diminished and there are new layers of elastic lamina: reduplication of elastic. The severity of renal lesions is correlated with the severity of luminal obstruction of arteries (Figures 5, 6, and 7).
Arterioles: The characteristic change is hyaline deposits in the wall of afferent arterioles: hyaline arteriolosclerosis. These hyaline deposits are initially seen in the subendothelial space and progressively increase in size until they occupy or replace the arteriolar walls. This material is very positive with PAS stain, is homogeneous, eosinophilic with H&E and with trichrome stains it can be seen red or green (or blue depending on the method used). The hyaline deposits are unevenly distributed, with arteriolar portions free of injury and other severely affected (Figure 8). According to the extension of hyaline changes in each arteriole and the number of arterioles with this change, hyaline arteriolosclerosis has been divided into three grades: Grade 1: small deposits in some arterioles, grade 2: large deposits in some arterioles or small deposits in many arterioles, and grade 3: extensive deposits in most arterioles (or mild, moderate and severe). Remember that if we identify hyalinosis in both arterioles (afferent and efferent) we must think in diabetic nephropathy.

Figure 5. Intrarenal artery with mild intimal fibrosis, this change is very common in adult patients, even without a diagnosis of hypertension. Asterisks indicate the muscle layer, the intima shows mild fibrosis (mild nefroangiosclerosis). (Masson's trichrome, X400).

Figure 6. Renal vascular changes in moderate hypertension. There is interstitial fibrosis with mild mononuclear inflammatory infiltrate, intimal fibrosis of arteries (green arrow) and hyaline arteriolosclerosis (blue arrows). (Masson's trichrome, X200).

Figure 7. Intrarenal artery with severe intimal fibrosis that obstructs its lumen approximately 80%. The arrows indicate the internal limit of the muscular layer. (Masson's trichrome, X400).

Figure 8. Renal tissue from a chronic hypertensive patient. In arterioles there are hyaline deposits in their walls (arrow). The severity of arteriolar hyaline changes has some degree of correlation with the time of evolution or severity of the systemic hypertension. (Masson's trichrome, X400).
Immunofluorescence
There is not specific deposits of immunoglobulins or complement, but arteriolar hyaline lesions and segmental glomerular lesions may have unspecific deposition (or trapping) of IgM and / or C3.
Electron microscopy
There are not specific findings. There are
some changes in glomerular basement membranes: thickening and irregularity;
and the hyaline deposits in arterioles are also identified.
MALIGNANT HYPERTENSION
The kidneys may show the changes of hypertension described above if the patient has a long history of hypertension, but when the disease is of recent onset these changes are not found. In the cortex and surface there are often hemorrhages and small petechiaes.
Histopathology
Arteries show a marked intimal thickening with mucoid or myxoid appearance, with myointimal cells. The internal elastic lamina is often attenuated, without fragmentation or reduplication. The lumen is significantly reduced. There may be intact or fragmented erythrocytes in the intima, and sometimes thrombosis. Small arteries and arterioles show areas with fibrinoid appearance in their wall, from the intima until the muscular layer: fibrinoid necrosis, and often thrombosis. Fibrinoid necrosis may extend from the arteriole to the glomerulus causing segmental or global tuft necrosis. It is also common to see intraglomerular capillary thrombosis (thrombotic microangiopathy). Other glomerular lesions that may be found are: mesangiolysis (pronounced dilation of capillaries with loss of mesangium), crescents and yuxtaglomerular hypertrophy. There are usually acute tubular necrosis and, occasionally, small cortical infarcts (Figures 9, 10 and 11).
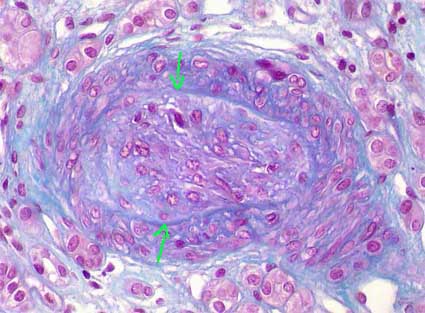
Figure 9. Severe obstruction of the lumen of an intrarenal artery, with proliferation of loose tissue and cells with large active nucleus, in a patient died due to acute brain damage in malignant hypertension. With H&E staining the myxoid appearance if intimal tissue is better seen. (Masson's trichrome, X400).

Figure 10. Renal arteriole from the same cases that the Figure 9. See the hyaline appearance in some segments of the wall, the presence of fibrinoid material (red) in the intima and fragmented erythrocytes (arrow). There is intraluminal thrombosis. Similar lesions can be seen in other causes of thrombotic microangiopathy. (Masson's trichrome, X400).

Figure 11. In advanced lesions of malignant hypertension there is concentric intimal fibrosis in small arteries and arterioles (green arrow). The glomerulus in this image shows segmental sclerosis (blue arrow) and the non sclerosed segment of the tuft shows solidification by increased mesangial matrix and decreased capillary lumina (red arrow). (Masson's trichrome, X400).
In chronic lesions the arteries show intimal thickening with concentric appearance ("onionskin") (Figure 11) and tubulointerstitial and glomerular chronic changes.
Lesions of the kidney in malignant hypertension are similar to that seen in scleroderma, which ultimately produces malignant hypertension.
With immunofluorescence we can detect fibrinogen,
IgM and C3 in fibrinoid lesions of arteries and arterioles. By electron microscopy
endothelial edema in arteries, arterioles and capillaries, with widening of
the subendothelial space, can be demonstrated.
.
It is a syndrome characterized by hypokalemia secondary to renal loss of potassium, metabolic alkalosis and a normal or reduced blood pressure. There are usually hypermagnesuria and hypomagnesemia. There is increased synthesis of renin and aldosterone, and possibly prostaglandin E2, prostacyclin, calicrein and bradykinin. Most cases are diagnosed in childhood. The disorder presents with polyuria and generalized weakness, attributed to hypokalemia. The disorder is usually inherited in an autosomal recessive manner, although sporadic cases are known. The physiopathogenic mechanism is not completely clear, but it seems that the key lies in an alteration in the reabsorption of chloride in the ascending branch of the thick loop of Henle. Inappropriate chloride reabsorption would produce volume depletion and would activate the renin-angiotensin system. The distal release of salt and water could promote secretion of potassium and hydrogen producing hypokalemia and alkalosis. Prostaglandin synthesis would increase due to volume contraction and hypokalemia. There may be that multiple physiopathogenic mechanisms exist.
A widely used system classifies Bartter syndrome on the basis of the underlying genetics, as follows (from: Frassetto LA. Bartter Syndrome, In eMedicine-Medscape: Link):
Type 1 – Antenatal Bartter syndrome: Results from mutations in SLC12A1, the sodium-chloride-potassium cotransporter gene.
Type 2 – Antenatal/neonatal Bartter syndrome: Results from mutations in the ROMK gene.
Type 3 – Classic Bartter syndrome: caused by mutations of the chloride voltage-gated channel Kb gene ( CLCNKB), which encodes the kidney-specific basolateral chloride channel (ClC-Kb) involved in sodium chloride reabsorption in the renal tubule.
Type 4 – Bartter syndrome with sensorineural deafness: Results from loss-of-function mutations in BSND, which encodes an essential beta subunit for chloride channels.
Type 5 – Gitelman syndrome: Results from mutations in SLC12A3, the sodium-chloride cotransportert.
The diagnosis is not based on histopathology, so a biopsy is not usually performed. The characteristic renal change is diffuse hypertrophy of the yuxtaglomerular apparatus and prominent medullary interstitial cells.
RENAL ARTERY FIBROMUSCULAR DYSPLASIA
The causes of renovascular disease are atherosclerosis, thrombosis, extrinsic compression, and stenosis of any cause. Ischemic renal disease occurs with an obstruction greater than 70% of the arterial lumen. Fibromuscular dysplasia is a rare cause of arterial stenosis; it is more frequent in women and predominantly affects young adults. It is classified based on the layer of the vessel affected:
- Intimal fibroplasia
- Medial hyperplasia
- Medial fibroplasia with aneurisms (the most common: 60-85%
of cases)
- Medial dissection: probably began in an area of intimal fibroplasia
- Perimedial fibroplasia
Stenosis of the main renal artery or its branches causes renovascular hypertension by activation of the renin-angiotensin-aldosterone system. Unilateral renal stenosis may cause changes of hypertensive nephropathy in the contralateral kidney; the kidney with stenosis is protected from the effects of hypertension, this phenomenon is known as the "Goldblatt kidney".
Fibromuscular dysplasia produces systemic hypertension and usually is asymptomatic in terms of renal injury until there are advanced lesions. The treatment is surgical and the prognosis depends on the extent of the lesions and the chronic renal damage occurred.
Histopathology
Renal parenchymal lesions are a consequence of ischemia due to the arterial lesions and the changes are similar to ischemia of any other cause. See histopathology in “Renal Involvement in Systemic Arterial Hypertension”. If the stenosis is unilateral, the kidney with stenotic lesion shows interstitial fibrosis and diffuse tubular atrophy, glomeruli are more clustered due to the atrophy of the cortical parenchyma among them, yuxtaglomerular apparatus can be hypertrophied and arteries are usually not thickened and they do not show hypertensive changes. Contralateral kidney (without stenotic lesion) has arterial and arteriolar sclerosis and other hypertensive changes.
Intimal fibroplasia: The intima appears thickened by a loose hypocellular fibrous tissue, the lumen is reduced and the internal elastic lamina is preserved. It is a very rare variant. Sometimes it can be found accompanying other forms of fibromuscular dysplasia.
Medial hyperplasia: There is thickening of the media with increased number of smooth muscle cells. No changes are found in the intima, internal elastic lamina or adventitia. It is also a rare variant. It often causes severe stenosis.
Medial fibroplasia with aneurysms: The most common form. There are areas of thickening and fibrosis in the media alternating with aneurysmatic dilatations where there is loss of elastic lamina. In areas where there is medial thickening the smooth muscle is replacement by collagen. It is often bilateral and usually does not cause a complete luminal obstruction.
Medial dissection: There are defects in the internal elastic lamina that provides access for blood to enter the media producing dissection. It is possible that this lesion begin in areas of intimal fibroplasia.
Fibroplasia perimedial:
There are accumulations of collagen replacing large areas of the muscular media,
mainly in the outer two thirds (near to the adventitia). The media appear irregularly
thickened.
It usually presents in patients with atheromatosis of abdominal or thoracic aorta and its major branches; the emboli usually occur after endovascular procedures or cardiac surgery. With some frequency we see cholesterol emboli in graft kidneys where the donor or the receptor has atheromatosis. Emboli may also occur spontaneously. Anticoagulants increase the likelihood of detachment of cholesterol emboli. These emboli can travel to any organ and produce diverse complications according to the organ and the severity of the obstruction. The kidneys are the most frequently involved for cholesterol emboli.
Clinic features:
Symptoms and signs are usually detected several days after the precipitating
event (usually surgery); skin and retinal vessels lesions can be seen. In kidneys
there is increase in BUN and serum creatinine. In urinalysis there may be proteinuria,
hematuria and casts. There may be also systemic blood pressure increase. In many cases there is eosinophilia and eosinophiluria. The
diagnosis can be demonstrated in skin biopsies. In many cases the emboli are
few, small and asymptomatic. In cases of renal dysfunction, some patients recover
function, but in others there is irreversible damage. There is not an effective
treatment for cholesterol thromboembolism.
The kidneys may show multiple infarcts and lesions in large vessels.
Histopathology
The distinctive and necessary finding for diagnosis is the presence of empty needle spaces in the lumen of arterioles, small arteries and occasionally arcuate arteries; these spaces are surrounded by fibrin thrombi and, if they have several days or weeks after they occurred, multinucleated giant cells (Figure 12). The spaces correspond to cholesterol crystals that dissolve with the normal processing of the tissue. To see these crystals is necessary to cut fresh frozen tissue and to see it with polarized light. Small emboli may reach glomerular capillaries and cause a gigantocellular reaction. In the first hours after the embolism there is infiltration of polymorphs that is after gradually replaced by lymphocytes and histiocytes. Depending on the involved artery we can see variable size infarcts. Surrounding ancient emboli a fibrous reaction develops.
See Case 188 of our Case Series: Cholesterol embolism.
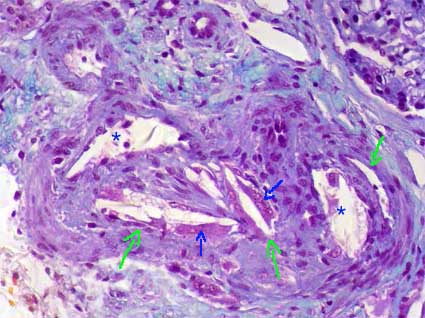
Figure 12. This intrarenal artery is distortion due to cholesterol crystals in its lumen. These crystals disappear with the processing and remain the space they occupied as needles with pointed ends (elongated diamond) (green arrows). They are surrounded by inflammatory cells, mainly multinucleated giant histiocytes (blue arrows), and partially or completely obstruct the lumen. Asterisks indicate residual lumen of the artery. (Masson's trichrome, X300).
It is an uncommon cause of renal failure in which there is severe cortical ischemia with relatively preserved medulla. It occurs when there is severe circulatory failure (shock) with or without lesions of renal arteries. The most common causes are obstetric complications such as abruptio placentae, eclampsia and septic abortion; but it is also seen in other instances such as haemolytic uremic syndrome, snake venom, intravascular hemolysis, severe graft rejection, and circulatory collapse of any case. The clinical presentation is that of an acute renal failure. Renal cortical necrosis is bilateral, although exceptionally it may be unilateral.
The kidneys have a dark surface and mottled appearance, the cortex is pale and the medulla congestive. Histologically, the cortex displays characteristics of an ischemic infarction that may be diffuse or in patches. Juxtamedullary cortex, medulla and a narrow rim of subcapsular cortical tissue (with irrigation for capsule vessels) are preserved, with congestive capillaries. There may be thrombus in arteries and arterioles. Eventually the necrotic areas tend to calcify.
HEMOLYTIC UREMIC SYNDROME AND THROMBOTIC THROMBOCYTOPENIC PURPURA
Hemolytic uremic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP) are closely related diseases and with very similar clinical manifestations; it is often difficult to obtain a clear differentiation between these two entities. Renal alterations are similar in both diseases. Lesions in renal arteries and capillaries are similar in HUS/TTP, eclampsia, malignant hypertension, scleroderma, acute humoral rejection and, sometimes, in cyclosporine toxicity, and are known as thrombotic microangiopathy.
Endothelial damage is key in developing HUS/TTP. The triggering factor for this injury may be bacterial endotoxins, antibodies against endothelial cells, an inducer of apoptosis, cyclosporine, mitomycin, and so on. Altered endothelial cells act as procoagulant and release Von Willebrand factor facilitating thrombosis in small vessels. A genetic predisposition or deficiency of an antiplatelet factor has also been implicated in the development of the disease. Hereditary forms of HUS and forms associated with abnormalities of the complement are described in many cases of HUS not associated to diarrhea.
The most common infections that trigger HUS/TTP are E. coli, Shigella disenteriae, Streptococcus pneumoniae, Salmonella typhi and viruses. Some cases have been associated with pregnancy, use of oral contraceptives, bone marrow transplantation, and radiation to the kidney. Many cases of TTP are considered idiopathic.
Clinical features: The HUS/TTP is a consumptive coagulopathy with microangiopathic hemolytic anemia, negative coombs, and thrombocytopenia with no other apparent cause. The patients also present fever, high blood pressure and kidney failure. The disease has a predilection for the kidney and central nervous system. The TTP occurs mainly in adults and HUS in children, especially those forms associated with diarrhea. Kidney failure is common in both diseases and is generally recognized by azoemia, macro or micro-hematuria, proteinuria and urine casts. Renal involvement is usually more severe in HUS; central nervous system is more frequently involved in TTP: impaired consciousness, focal signs, and so on.
The Coombs test is negative (it is not an autoimmune hemolysis), the erythrocyte present changes related to the microangiopathy: fragmentation and schistocytes. There is no alteration in levels of serum complement. To make the diagnosis of HUS/TTP a renal biopsy is not required.
The HUS has been divided into two variants, one associated to diarrhea: Shiga toxin-producing E. coli-associated hemolytic uremic syndrome (STEC-HUS) (formerly classic HUS or HUS diarrhea positive or HUS D+), and the other one non-associated to diarrhea: atypical HUS.
STEC-HUS: This form, also called the classical form, presents after or during an episode of diarrheal disease, there is acute renal failure with oliguria, proteinuria, hematuria, anemia and thrombocytopenia. D+ HUS is the classic form, accounting for 95% of cases of hemolytic uremic syndrome in children. It may or may not be associated with systemic manifestations. We can find skin petechiae, purpura, intestinal bleeding, systemic hypertension, and neurological disorders: convulsions, aphasia, visual alterations, coma, and so on. In these cases microangiopathy is more often evidenced in the glomeruli. Episodes are frequently related to "endemic outbreaks”, mainly in summer. The prognosis is better in this form of HUS.
Atypical SHU: Can occur in children or adults. There is not diarrhea preceding or during the clinical episode. Renal failure and systemic hypertension are more severe. aHUS has been the focus of current research and has, thus far, been associated with complement dysregulation in up to 50% of cases. Specifically, mutations in complement regulatory protein factor H, factor I, or factor B or autoantibodies against factor H have all been implicated. These mutations result in inability to suppress complement activation and for reasons that are not completely understood, the glomerular endothelium is particularly susceptible to these changes. D- HUS has been associated with various nonenteric infections, viruses, drugs, malignancies, transplantation, pregnancy, and other underlying medical conditions such as scleroderma and antiphospholipid syndrome. Infections caused by Streptococcus pneumoniae has been linked to 40% of aHUS cases. Categories of drugs that have been most frequently associated with D- HUS include anticancer molecules (mitomycin, cisplatin, bleomycin, and gemcitabine), immunotherapeutics (cyclosporine, tacrolimus, OKT3, IFN, and quinidine), and antiplatelet agents (ticlopidine and clopidogrel) (Tan AJ. Hemolytic Uremic Syndrome. In E-Medicine [Link]). At least 50% of patients with aHUS have an underlying inherited and/or acquired complement abnormality, which leads to dysregulated activity of the alternative pathway at the endothelial cell surface. There are, however, noncomplement inherited abnormalities such as mutations in DGKE, which can result in an aHUS phenotype. Atypical hemolytic uremic syndrome is a “thrombotic” microangiopathy (TMA), the pathological features of which represent tissue responses to endothelial injury. In some biopsies, overt thrombosis as evidenced by intraluminal fibrin or fibrin-platelet plugging is not seen. Nonthrombotic features include endothelial swelling and denudation, mesangiolysis, double contours of the glomerular basement membrane, and subendothelial accumulation of electron-lucent, flocculent material. In arteries and arterioles, intramural fibrin, myxoid intimal thickening, and concentric myointimal proliferation (onion-skinning) may occur (Goodship TH, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int. 2017;91(3):539-551. [PubMed link]).
Some clinicians now use the term “primary aHUS” when an underlying abnormality of the alternative pathway of complement is strongly suspected and other causes of secondary aHUS have been excluded. However, even in some of these patients, a complement abnormality will not be identified. In many patients with an underlying complement risk factor, a trigger is required for aHUS to manifest. Triggers include autoimmune conditions, transplants, pregnancy, infections, drugs, and metabolic conditions. It may be difficult to show unequivocally that a trigger unmasks latent complement defects. Additional work is required to define the impact of complement risk factors in these subgroups. Following the diagnosis of a TMA, clinical and laboratory evaluation is required to establish the etiology. ADAMTS13 activity is urgently required to exclude TTP prior to treatment with eculizumab in adults but is not a prerequisite in children. Investigation for STEC-HUS should be undertaken in all individuals with suspected aHUS. In all pediatric aHUS, plasma and urinary evaluation for cblC deficiency is mandatory. All individuals with suspected primary aHUS should have a complete evaluation for complement-mediated aHUS (Goodship TH, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int. 2017;91(3):539-551. [PubMed link])..
The prognosis of the disease is usually good. Mortality in TTP is near to 20%; in STEC-HUS is near to 4%, and in aHUS is near to 10%. HUS in adults is more heterogeneous in its presentation and mortality is up to 30%. HUS cases in endemic outbreaks have a better prognosis. The severity of systemic hypertension and duration of anuria are also factors related to prognosis. In up to 39% of cases there is residual renal damage after HUS (Kern WF, Silva GF, et al, Atlas of Renal Pathology. WB Saunders Company, Philadelphia, 1999, p. 69). For treatment corticosteroids, antibodies, plasma exchange, renal failure control, and other supportive measures are used. Eculizumab is the first treatment approved by the US Food and Drug Administration (FDA) (September, 2011) for adults and children with atypical hemolytic uremic syndrome.
Macroscopically the kidneys present subcapsular petechiae, and frequently there are areas with necrosis. In cases of a prolonged disease the kidneys appear decreased in size and there is narrowing of arterial lumina.
Histopathology
Glomerular capillary walls appear swollen due to subendothelial edema, often giving a double contour appearance due to separation of the endothelium and the GBM, or due to neoformation of a new “GBM” between endothelial cells and the true GBM; between these two “GBM” there may be “interposed” mesangial cells. In capillary lumina there are microthrombi with fibrin, platelets and trapped fragmented erythrocytes. These thrombi can completely fill the capillary and may adhere to the capillary wall without completely occlude the lumen. When there is severe vascular lesions the glomeruli appear distended and with severe congestion: "glomerular paralysis”, and in severe cases there will be necrosis of the tuft. Sometimes there are accumulations of polymorphs. The mesangium is widened and in some cases appears swollen or with fibrillary appearance, without hypercellularity. We can also see meangiolysis characterized by loss of matrix and mesangial cells with capillaries that appear expanded and loosed. In a few cases there are crescents, usually few and circumscribed. In severe vascular lesions glomeruli appear with ischemic changes.
In advanced lesions (or chronic) there is
increased mesangial matrix (mesangial sclerosis) with thickened capillary walls
with double contours; we can see also ischemic changes. Double contours may
persist for several years, but in general, the glomeruli do not show similar
features to the membranoproliferative GN since generally there is no conspicuous
hypercellularity (Figure 13). In cases of thrombotic microangiopathy associated
with immune complexes diseases, such as lupus, we can see glomerular hypercellularity
and double contours that can be similar to idiopathic type I membranoproliferative
GN.

Figure 13. Glomerular thrombotic microangiopathy in a patient with several weeks of evolution. In this glomerulus there are not thrombi, but capillary walls are thickened by edema (arrows) with decreased capillary lights, the tuft appears more "solid" and without hypercellularity. Wuth methenamine-silver double were observed. (Masson's trichrome, X400).
In tubules is common to see changes of NTA, erythrocyte casts and calcifications. In the interstitium there is edema and mild mononuclear inflammatory infiltrate. In severe cases we will find areas of necrosis and / or hemorrhage.
Arteries: In some cases they appear normal; in early stages there is endothelial edema. Fibrinoid necrosis is commonly found in the arteriolar wall, without inflammatory cells, accompanied by arteriolar thrombi and fragmented red cells in the wall (Figure 10). These thrombi can extend to glomeruli producing global or segmental necrosis. In some arterioles there are dilatations (aneurysms), usually near the glomerulus; it is said that this injury is more common in PTT (Kern WF, Silva GF, et al, Atlas of Renal Pathology. WB Saunders Company, Philadelphia, 1999, p. 71). In cases of organized thrombus, the recanalization may take the appearance of tufts, wherefore authors call these organized thrombi “glomeruloid srtructures”. In lesions of more time evolution, the intima is thickened with growth of cells (myointimals), and often adopts a pattern of concentric thickening similar to that seen in malignant hypertension "onion skin"; in fact, many of the histological alterations in the SHU / PTT are similar to those of malignant hypertension. The residual lesion is intimal fibrosis, difficult or impossible to distinguish from any other cause.
Immunofluorescence
There is fibrin or fibrinogen in glomerular capillary walls and arteriolar lesions. We can also detect IgM, C3, IgG and IgA in glomerular capillary walls. In arterioles there is IgM and, sometimes, C3, C1q and other Igs. These deposits of Igs. and complement fractions are nonspecific, probably not pathogenic.
Electron microscopy
There is edema in the subendothelial space
(lamina rara interna) and endothelial cells. It is common to see interposition
of mesangial cell between GBM and endothelial cell. The subendothelial space
is pale, acellular and with fibrillar or granular appearance. There are areas
of detachment of endothelial cells. The mesangium has a finely fibrillar or
granular appearance.
There is detachment of the podocyte processes and deposits of material similar
to the GBM in its internal aspect (between GBM and endothelium or between the
interposed mesangial cell and endothelium); this is the material originating
the double contour. Furthermore, there are arteriolar and capillary thrombi,
and the arteriolar wall lesions that were described in histopathology.
See Case 97 of our case series: HUS
See Case 177 of our Case Series: HUS
Preeclampsia is characterized by hypertension, proteinuria and edema in the third trimester of pregnancy. Eclampsia is defined by the presence of seizures in a patient with signs of preeclampsia. The clinical features are not specific and can occur in various renal or extrarenal diseases, that are accentuated by underlying physiological changes of pregnancy. Renal biopsy is rarely performed in these patients.
Histological changes are predominantly glomerular and resemble those of HUS / TTP. There are marked edema and endothelial cell hypertrophy: "endotheliosis". There is also vacuolization of mesangial and endothelial cells, increased mesangial matrix, usually without or with slight mesangial hypercellularity; sometimes mesangial interposition and double contour of the capillary walls (most common in severe cases or in the resolution phase) are evidenced. It's not often find glomerular or arterial thrombosis (Figures 14 and 15). In arteries and arterioles we can see nonspecific changes as intimal fibrosis, medial hypertrophy and arteriolar hyaline deposits; there is usually no fibrinoid necrosis.
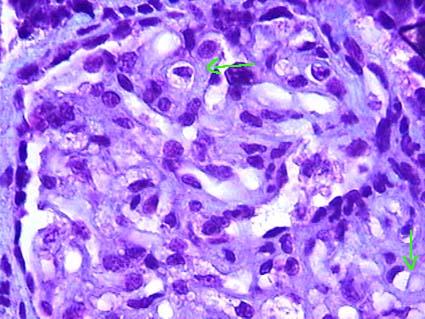
Figure 14. Glomerulus from a patient who died from eclampsia with HELLP syndrome; they were evaluated for possible kidney transplant. Both were transplanted and, despite having delayed graft function, after a week they started to function. See this glomerulus with marked reduction of capillary lumen, thick walls (arrows) and prominent endothelial cells (endotheliosis). The glomerulus appears without elements of the blood (anemic). (Masson's trichrome, X400).

Figure 15. This glomeruls is from the same case of the previous figure. There is a capillary thrombi (green arrow), hypertrophy and hyperplasy of podocytes, one of them in mitosis (blue arrow) and capillary lumina narrowed (Masson's trichrome stain, X400).
Immunofluorescence shows similar findings of HUS / TTP and on electron microscopy we can see also, according to the severity of the injuries, some findings as described in HUS / TTP.
The prognosis is excellent if the patient receive timely and appropriate medical care. Symptoms and signs resolve rapidly after termination of pregnancy. The histological lesions also disappear in a few days, usually without morphological sequelae.
LARGE AND MEDIUM VESSEL VASCULITIDES
In the Extracapillary Glomerulonephritis chapter we discussed about small vessel vasculitis involving the kidney: Granulomatosis with polyangiitis (formerly Wegener’s granulomatosis), microscopic polyangiitis and eosinophilic granulomatosis with polyangiitis (Churg-Strauss). In addition, in other chapters discussed about Henoch-Schönlein and cryoglobulinemia, which can be considered other types of vasculitis. Here we refer to the vasculitis of large and medium vessels. Of these, the most frequently affecting the kidney is polyarteritis nodosa, the others rarely do.
Vasculitides can be defined, in the simplest manner, as inflammation of the vascular wall. The kidney is affected in many of them, sometimes being the most affected organ and sometimes as a minor injury that may go unnoticed.
The most accepted nomenclature in vasculitides is the 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides, a reviewed version of the 1994 Conference (Jennette JC, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013 Jan;65(1):1-11. [PubMed link]):
Large vessel vasculitis
- Takayasu arteritis
- Giant cell arteritis
Medium vessel vasculitis
- Polyarteritis nodosa
- Kawasaki disease
Small vessel vasculitis
- Antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis
- - Microscopic polyangiitis
- - Granulomatosis with polyangiitis (Wegener’s)
- - Eosinophilic granulomatosis with polyangiitis (Churg-Strauss)
- Immune complex small vessel vasculitis
- - Anti–glomerular basement membrane (anti-GBM) disease
- - Cryoglobulinemic vasculitis
- - IgA vasculitis (Henoch-Schönlein)
- - Hypocomplementemic urticarial vasculitis (anti-C1q vasculitis)
Variable vessel vasculitis
- Behcet’s disease
- Cogan’s syndrome
Single-organ vasculitis
- Cutaneous leukocytoclastic angiitis
- Cutaneous arteritis
- Primary central nervous system vasculitis
- Isolated aortitis
- Others
Vasculitis associated with systemic disease
- Lupus vasculitis
- Rheumatoid vasculitis
- Sarcoid vasculitis
- Others
Vasculitis associated with probable etiology
- Hepatitis C virus–associated cryoglobulinemic vasculitis
- Hepatitis B virus–associated vasculitis
- Syphilis-associated aortitis
- Drug-associated immune complex vasculitis
- Drug-associated ANCA-associated vasculitis
- Cancer-associated vasculitis
- Others
Another approach to classification is by its
etiology (humoral autoimmunity, pauciimmune, infectious, cell injury mediated).
Importantly, according to the classification of Chapel Hill, the presence of
small vessel lesions is incompatible with the diagnosis of medium vessels vasculitis;
however, sometimes we can find medium-sized artery lesions in small vessel vasculitis.
And remember that the glomeruli are capillaries, i.e. small vessels. In other
words, if there is GN is a small vessel vasculitis.
The small vessel vasculitides have been discussed in other chapters, now review
the kidney involvement in large and medium vessels vasculitides.
Large vessel vasculitis
In these diseases there is rarely renal involvement. Lesions can be identified in the main renal artery and/or in intraparenchymal arteries. Giant cell arteritis and Takayasu arteritis are characterized by granulomatous inflammation of the arterial wall, most prominent in the media, although there is lympho-histiocytic infiltrate across the wall (panarteritis). In the active phase there are granulomas, giant cells and fragmentation of the internal elastic lamina; occasionally there is fibrinoid necrosis, but in these cases we should think in other types of vasculitis.
The distinction between the two types of large vessel vasculitis is clinical more than histological. There are reports of large-vessel arteritis with glomerular involvement, but, according to current concepts, we should think that they are two different processes.
Medium vessel vasculitis
Polyarteritis nodosa (PAN): Vascular lesions are localized in medium-sized vessels: interlobar, arcuate and interlobular arteries. In the same biopsy it is frequent to find lesions in different stage: acute and chronic. In the active phase there is fibrinoid necrosis in segments of the artery, with leukocyte infiltration; in early phases predominate neutrophils which fragment in the arterial wall: leukocytoclasia, subsequently neutrophils are replaced by lymphocytes, monocytes and macrophages. The necrosis begins on the inside of the artery wall and can be transmural, affecting non continuous sections, so that sometimes it may not be easy to find; it is most often located at the bifurcation of the arteries. Fibrinoid necrosis is eosinophilic with H&E and red with trichrome stains and is accompanied by karyorrhexis and destruction of the internal elastic lamina (Figure 16). We can detect thrombi in areas of necrosis, which can lead to cortical infarctions that appear triangular. Nodules that give the name to the disease are areas of vasculitis with inflammation around, they are more frequently seen in the skin, but they can be seen in other organs such as the kidney. In some cases there are aneurysms due to arterial wall destruction (also originate nodules). In the chronic stages of injury there is vascular sclerosis, recanalization of thrombus and organization of the injured tissue, this can lead to renovascular hypertension. In cases where we only found adventitial inflammation we cannot diagnose PAN, it is necessary to demonstrate the acute lesions of vasculitis.
[See image of artery with fibrinoid necrosis in PAN (link)] [Another image (link)]
[Go to Case 108 of our case series]

Figure 16. Cortical radial artery with fibrinoid necrosis of its wall (blue arrows) and inflammatory cells in the middle of this fuschinofílic material (red). Green arrows indicate arterial wall segments without lesion and asterisks the vessel lumen (Gomori’s trichrome stain, X400).

Figure 17. Medium-sized artery with fibrinoid necrosis of the wall, karyorrhexis and thrombosis partially occluding its light, this case corresponds to a patient with polyarteritis nodosa (H&E, X200).
Kawasaki disease: is arteritis associated with the mucocutaneous lymph node syndrome and predominantly affecting medium and small arteries. Coronary arteries are often involved; aorta and large arteries may be involved. Usually occurs in infants and young children. It is characterized by fever, lymphadenopathy and inflammation of mucous membranes, skin and arteries: mainly coronary, axillary and iliac. In approximately 25% of cases there is involvement of the main renal artery, but rarely renal dysfunction occurs. Histologically, in arteries, there is edema, wall infiltration by neutrophils and mononuclear cells, and perivascular inflammation. By evolving the process reduces the infiltration of neutrophils and lympho-histiocytic persists. There is less arterial necrosis than in PAN; in later stages of the process there is intimal fibrosis of varying degree. Occasionally has been describes intrarrenal artery involvement..
Bibliography
Hypertensive nephropathy
HUS/TTP
Preeclampsia - Eclampsia
Large and medium vessel vasculitides
Homepage - - - Tutorial Index