
Neoplasias renales
Parte 1. Generalidades - Clasificación - Carcinoma de células renales (CCR) - Estadificación - CCR convencional - Neoplasia renal quística multilocular de bajo potencial maligno - CCR papilar - CCR cromófobo - Carcinoma de conductos colectores - Carcinoma medular - Carcinoma de conductos colectores de bajo grado - Carcinoma tubuloquístico
Parte 2. Carcinoma mucinoso tubular y de células fusiformes - CCR asociado a la translocación Xp11.2 - CCR inclasificable y CCR en enfermedad renal quística adquirida - Adenoma papilar - Oncocitoma - Diagnóstico diferencial en tumores con células granulares - Adenoma metanéfrico - Adenofibroma metanéfrico - Tumor estromal metanéfrico - Angiomiolipoma - Tumor renomedular de células intersticiales
Parte 3. Tumor de células yuxtaglomerulares - Nefroma quístico - Tumor estromal y epitelial mixto - Sarcoma sinovial - Nefroblastoma (tumor de Wilms) - Nefroblastoma quístico parcialmente diferenciado - Restos nefrogénicos (y nefroblastomatosis) - Sarcoma de células claras - Tumor rabdoide - Nefroma mesoblástico congénito - Tumor renal osificante de la infancia
Parte 2
Carcinoma mucinoso tubular y de células fusiformes
También se le ha llamado: “neoplasia epitelial renal mixoide de bajo grado” (“Low-grade myxoid renal epithelial neoplasm”), “carcinoma renal de células cúbicas y fusiformes" ("spindle and cuboidal renal cell carcinoma”) y "carcinoma de células fusiformes de bajo grado" (“Low-grade spindle cell carcinoma”). Macroscópicamente son lesiones sólidas, bien definidas y, usualmente, sin necrosis.
Hallazgos microscópicos: Es una lesión con características citológicas de bajo grado (grado 2 de Fuhrman) que mezcla áreas con aspecto tubular y sólido en medio de un estroma mixoide, con áreas sólidas fusocelulares. Previamente se clasificaba como "inclasificable" o "sarcomatoide" (aunque no es en realidad un cambio sarcomatoide, que implica alto grado). Las áreas tubulares presentan células cúbicas, con citoplasma débilmente eosinofílico, núcleos redondos, homogéneos, con nucléolo pequeño o no visible, poco pleomorfismo y escasas mitosis. Hay un estroma de aspecto mixoide o mucionoso (puede ser focal) que tiñe positivo con tinciones para mucinas ácidas. Las áreas fusocelulares muestran células con citoplasma y núcleos elongados, aspecto homogéneo, sin anaplasia, no hay características verdaderamente sarcomatoides, y escasas o ninguna mitosis identificable; estas áreas pueden ser similares a un leiomioma o a un sarcoma de bajo grado. La lesión es poco infiltrativa y no hay desmoplasia. En algunos casos hay áreas de necrosis focal, pero este hallazgo no se correlaciona con el comportamiento clínico del tumor, al menos en nuestros casos.
Figura 14. Carcinoma mucinoso tubular y de células fusiformes. Las células cúbicas se disponen formando túbulos irregulares en medio de un estroma con aspecto mucoide, basófilo (mixoide). Estas áreas se mezclan con áreas fusiformes. El grado nuclear suele ser 2 de Fuhrman, hay pocas mitosis y, en algunos casos, hay necrosis. Son lesiones poco metastatizantes. (H&E, X300).

Figura 15. Carcinoma mucinoso tubular y de células fusiformes. En este campo podemos ver áreas fusocelulares con carcaterísticas nucleares de bajo grado. Es muy importante no confundir este aspecto con el de carcinomas sarcomatoides: lesiones de alto grado y peor pronóstico.(H&E, X300).
Algunos autores han propuesto que se trata de lesiones originadas en, o diferenciadas hacia, células de la parte distal de la nefrona o del asa de Henle, sin embargo, otros trabajos, incluyendo trabajos nuestros no publicados aún, no respladan esta hipótesis.
Por inmunohistoquímica hay positividad para CK7, AE1/AE3 (coctel de CK) y CAM 5.2; negatividad para CEA, CD15, CD10, villina (estos tres últimos son marcadores de parte proximal de la nefrona), CK20 y RCC. Hay expresión variable de S-100, EMA, vimentina, Ulex europaeus, CK8, CK18 y CK19. Otros autores informan positividad para CD15 en muchos de los casos (Shen SS, et al. Ann Diagn Pathol. 2007;11:13-21 [PubMed link]) Con el marcador Ki67 (MIB 1) el índice de proliferación celular es usualmente < 1%. La expresión de marcadores por IHQ es similar en células cúbicas y fusiformes, comprobando el mismo origen y diferenciación en ambos tipos de células, a pesar de su aspecto citológico diferente.
Por microscopía electrónica se evidencian, en las células fusiformes, desmosomas, borde microvelloso, borde luminal y tonofilamentos; estos hallazgos también confirman el origen epitelial de estas células.
Citogenética: Con FISH e hibridación genómica se encuentra combinación de pérdidas cromosómicas de 1, 4, 6, 8, 13, 14 y ganancias de 7, 11, 16, 17, sin identificarse un patrón citogenético característico del tumor como en el convencional y el papilar.
Este tumor tiene un comportamiento poco agresivo; hasta el presente sólo se ha documentado metástasis en un caso (aproximadamente el 1%). En nuestra serie, este tipo corresponde a poco menos del 1% de carcinomas renales y en ningún caso se ha documentado metástasis o muerte del paciente debido al tumor.
Carcinoma renal asociado con la translocación Xp11 (ó Xp11.2) y traslocación 6;11 / fusión del gen TFE3 o del gen TFEB (denominados en la clasificación 2016 de la OMS: Carcinoma de células renales con translocación de la familia MiT)
En estos carcinomas hay fusión de genes involucrando dos miembros de la familia de factores de transcripción MiT: TFE3 and TFEB. Los carcinomas asociados con traslocación Xp11 presentan fusión de genes involucrando TFE3, y los que se asocian con (6;11) presentan una fusión de los genes MALAT1-TFEB
En la clasificación de la OMS de 2022, se incluyen los siguientes tipos de carcinomas "definidos molecularmente": 1.) Carcinomas de células renales con reordenamiento TFE3 - 2.) Carcinomas de células renales con TFEB alterado - 3.) Carcinoma de células renales con ELOC mutado (antes TCEB1) - 4.) Carcinoma de células renales con deficiencia de fumarato hydratasa - 5.) Carcinoma de células renales deficiente en succinato deshidrogenasa (WHO Classification of Tumours Editorial Board. Urinary and male genital tumours [Internet]. Lyon (France): International Agency for Research on Cancer; 2022. (WHO classification of tumours series, 5th ed.; vol. 8). Available from: https://tumourclassification.iarc.who.int/chapters/36.). Ver la tabla completa en Neoplasias parte 1.
El TFE3 codifica un factor de transcripción nuclear; puede fusionarse con otros genes: fusión PRCC-TFE3; fusión ASPL-TFE3; y fusión PSF-TFE3. La fusión ASPL-TFE3 es similar a la descrita en el sarcoma alveolar de tejidos blandos (en este tumor también hay expresión nuclear de la aproteína TFE3). La fusión ASPL-TFE3 se relaciona con estadios avanzados y metástasis linfáticas.
Es un tumor predominantemente de jóvenes (usualmente >10 años) y adultos jóvenes, aunque se describen casos en mayores de 50 años. Se suelen diagnosticar en estadios avanzados debido a gran tamaño tumoral y extensión extrarrenal, pero no suelen detectarse metástasis. Hasta ahora parece un tumor poco frecuente (nosotros hemos diagnosticado 2 casos entre más de 500 tumores renales). A pesar de su rápido crecimiento local y del alto estadio inicial, parece que no son muy metastatizantes, aunque debemos esperar estudios más amplios para conocer mejor su comportameinto clínico.
Macroscópicamente tienen un aspecto similar al CCR convencional.
Hallazgos microscópicos: hay un aspecto variable, con áreas parecidas o similares al carcinoma convencional, pero suelen tener un aspecto más variable y muchas veces se ven con células más grandes, voluminosas, irregulares, como si viesemos un carcinoma convencional "raro", con carcaterísticas singulares. En muchos hay células claras, aspecto papilar o en nidos; podemos ver microcalcificaciones. En otros casos las células grandes y voluminosas tienen citoplasma granular eosinofílico. Los núcleos son redondos y usualmente grado 2 y 3. El número de mitosis es variable, en nuestros casos pocas (entre 1 y 3 por 10 campos de gran aumento). Hay poca anaplasia. Se dice que los tumores con fusión ASPL-TFE3 tienen un aspecto más papilar y los que tienen la fusión PRCC-TFE3 un patrón más compacto.

Figura 16. Carcinoma renal en una mujer de 17 años; masa que reemplaza gran parte del órgano y se extiende al retroperitoneo. En este caso el aspecto recuerda el de carcinomas con células granulares. El estudio de inmunohistoquímica (realizado en otro centro) fue positivo para TFE3. (H&E, X400).uerda

Figura 17. Carcinoma renal asociado a la translocación Xp11.2 en una mujer de 19 años, su aspecto es de células claras, voluminosas, con áreas papilares y múltiples microcalcificaciones. (H&E, X300).
Por IHQ lo más carcaterístico es la inmunomarcación nuclear para TFE3. Hay positividad para RCC y CD10. EMA y CK se detectan en aproximadamente 50% de casos.
Para efectos prácticos, debemos pensar en este tumor en pacientes jóvenes (desde niños hasta cuarta década de la vida), con un aspecto histológico que recuerda en algo al carcinoma convencional, pero, con carcaterísticas extrañas o poco usuales y además gran tamaño. Sin embargo, recordemos que el aspecto clínico, macro y microscópico es variable, así que es posible que en muchos tumores encontremos esta translocación, por lo que debemos esperar más estudios para conocer más acerca de este tumor. (Salles PG, Soto M Jr. Kidney carcinoma associated with Xp11.2 translocation / TFE3 (ASPL-TFE3) gene fusion. Int Braz J Urol 31:251-4, 2005. [PubMed link / Free full text]).
Carcinoma de células renales inclasificable
No es un tipo específico de carcinoma; aquí se agrupan los carcinomas renales que no encajan dentro de las restantes categorías. Corresponde a menos del 1% de carcinomas renales. No tienen un aspecto morfológico distintivo y su pronóstico no puede determinarse con certeza ya que reúne un grupo heterogéneo de tumores.
Esta categoría debería reservarse para casos con: 1.) Histología diferente a cualquiera de los otros subtipos; 2.) extensa necrosis con mínimo tumor viable que hacen imposible determinar el tipo histológico; 3.) tumores mucosecretores; 4.) carcinomas extensamente sarcomatoides sin elementos epiteliales reconocibles; y 5.) mezcla de tipos histológicos conocidos de carcinomas, o carcinomas con componentes estromales.

Figura 18. Carcinoma renal en gran parte sólido, con extensa necrosis y sin las carcaterísticas de ninguno de los otros carcinomas descritos en la clasificación de tumores renales. Fue diagnosticado como carcinoma renal inclasificable. (H&E, X300).

Figura 19. Otro tumor renal sin un patrón característico. La categoría "inclasificable" se aplica en estos casos o cuando hay mezcla de tumores conocidos, cambio sarcomatoide difuso, necrosis generalizada que hace imposible determinar el tipo o tumores mucosecretores. (H&E, X300).
Carcinoma de células renales en enfermedad renal quística adquirida
La principal complicación de la enfermedad renal quística adquirida (ERQA) (la cual se presenta en pacientes en diálisis crónica y enfermedad renal terminal) es el desarrollo frecuente de tumores renales, incluido el carcinoma de células renales (CCR).
El CCR de células claras es el tipo histológico más común en pacientes con enfermedad renal terminal (ERT), independientemente de la duración de la hemodiálisis. Los CCR tienden a mostrar cambios sarcomatoides durante la hemodiálisis de muy larga evolución, especialmente cuando supera los 20 años. En conjunto, estos hallazgos sugieren que las toxicidades asociadas a la hemodiálisis, como las que causan estrés oxidativo continuo a largo plazo, provocan numerosos cambios genéticos que promueven el desarrollo de CCR, incluida la transformación sarcomatoide, en pacientes con ERT. Los CCR en pacientes con ERT se describieron por primera vez en 1977, las características histológicas de los seis CCR en ese estudio parecían similares a las de los CCR de células claras y papilares. El CCR asociado a la ERQA fue categorizado como una nueva entidad por la OMS en 2016. Los CCR asociados a la ERQA generalmente ocurren dentro de un quiste en un contexto de múltiples quistes. A veces son multifocales y bilaterales, y generalmente están bien circunscritos, excepto en casos con componente sarcomatoide. La superficie de corte es típicamente de color ocre a amarillo o marrón y a veces se ven hemorrágicos y necróticos.
La mayoría de los CCR asociados a la ERQA tienen múltiples patrones histológicos (alveolar, tubular, papilar, quístico y sólido) en diferentes proporciones. Muchos tienen arquitectura cribiforme. Las células tumorales generalmente tienen abundante citoplasma eosinofílico y núcleos redondos a ovales con nucleolos prominentes. Hay pocas figuras mitóticas. La presencia de cristales de oxalato de calcio que muestran birrefringencia multicolor bajo luz polarizada es una de las características histológicas más distintivas del CCR asociado con ERQA. Los cristales de oxalato de calcio a menudo se observan tanto en el estroma como en el tumor y, curiosamente, a veces se asocian con células gigantes multinucleadas como resultado de reacción de tipo cuerpo extraño. Su reconocimiento ayuda a diagnosticar CCR asociado a ERQA. Inmunohistoquímicamente, las células tumorales de CCR asociados a ERQA expresan marcadores de CCR, CD10, AMACR, glutatión S-transferasa-alfa y BerEP4, pero no citoqueratina 7, E-cadherina, citoqueratina de alto peso molecular o MOC31. Los estudios citogenéticos muestran ganancia de cromosomas 3, 7, 12, 16, 17, 20 e Y. Los quistes están recubiertos por células atípicas con citoplasma eosinofílico y tienen patrones de proliferación plana, papilar o cribiforme y anomalías citogenéticas, incluida ganancia de cromosomas. 3, 7, 12, 16, 17, 20 e Y. Aunque la proliferación celular atípica podría indicar premalignidad, como lo sugieren algunos estudios, no hay evidencia directa de que los CCR asociados a ERQA surjan de quistes. (La mayor parte del texto anterior fue tomado de: Tsuzuki T, et al. Renal tumors in end-stage renal disease: A comprehensive review. Int J Urol. 2018;25(9):780-786. [PubMed link]).
Ver Caso 167 de nuestra serie de casos: Carcinoma renal con depósitos de oxalato de calcio en paciente con enfermedad renal quística adquirida.
La clasificación de la OMS de 2022 los define así: "Los adenomas papilares son tumores no encapsulados con una arquitectura papilar o tubular de grado bajo y un diámetro igual o menor a 15 mm". El diagnóstico de adenoma papilar basado en una biopsia con aguja debe realizarse con extrema precaución, ya que la presencia de cualquier cápsula o problemas de heterogeneidad de grado puede no visualizarse" (Moch H, et al (Eds): WHO Classification of Tumours of the Urinary System and Male Genital Organs (4th edition). IARC: Lyon 2016; p 42).
Anteriormente, de acuerdo a su tamaño y morfología, se habían dividido en las siguientes categorías:
Claramente benignos:
< 0.5 cm con arquitectura túbulo-papilar
Malignos: > 1 cm
Probablemente malignos: < 1 cm pero con crecimiento
sólido o con células claras
Probablemente benignos: Igual que los “claramente
benignos” pero hasta 1 cm
Son asintomáticos y se descubren como hallazgo incidental; en autopsias
se encuentran entre el 4 y 37%; su frecuencia aumenta progresivamente con la
edad, siendo cercana al 40% en mayores de 70 años. Con el aumento del
uso de estudios de imágen de alta resolución cada vez se descubren
más como hallazgo incidental en vivos. Se asocian con enfermedad renal
terminal, hipertensión arterial, diálisis y enfermedad quística
adquirida. Los adenomas pueden coexistir con carcinomas renales de diferentes
tipos histológicos.
Hallazgos macroscópicos: Son lesiones bien definidas, aperecen como puntos amarillos a grisáceos, usualmente subcapsulares; siempre corticales.
Hallazgos microscópicos: Son lesiones papilares, tubulares o tubulopapilares; formadas por células con citoplasma escaso, anfófilo o basófilo, núcleos redondos u ovales con cromatina densa y nucléolo no distinguible; pueden haber vacuolas o hendiduras nucleares, no hay pleomorfismo y son inusuales las figuras mitóticas. No deben haber células claras, de lo contrario debemos pensar en carcinoma.
Citogenetica: Se han descrito ganancias en los cromosomas 7, 12, 16, 17 y 20, y pérdida del cromosoma Y; estas aletarciones sugieren una estrecha relación (o similitud) con el carcinoma papilar. No encontramos alteraciones en 3p.
Una lesión relacionada con el adenoma es la hiperplasia epitelial renal; está formada por acúmulos compactos de células epiteliales uniformes que adopatan un patrón tubular o papilar y son múltiples, coticales, uni o bilaterales; no son visibles macroscópicamente. Se asocian con adenoma papilar.
Es un tipo característico de tumor benigno, separado de los tumores restantes desde los años setenta. Algunas diferencias en los criterios para diagnosticarlo han llevado a casos controvertidos de "oncocitoma con metástasis", "oncocitoma atípico" y "oncocitomas grado 2". Cuando es diagnosticado con criterios morfológicos estrictos es una lesión claramente benigna. Corresponde a aproximadamente el 5% de neoplasias epiteliales de adultos. Se postula que se originan de las células intercaladas de conductos colectores (igual se dice de los carcinomas cromófobos).
Son lesiones usualmente asintomáticas; algunas veces se manifiestan con dolor, masa abdominal o hematuria.La edad media al diagnóstico es aproximadamente 62 años, con un rango bastante amplio: 10 - 94 años. Es más frecuente en hombres con una relación H:M de 2:1. Se describen cuatro carcaterísticas angiográficas que sugieren su diagnóstico: 1.) un anillo lúcido creado por la cápsula tumoral; 2.) un patrón capilar homogéneo con densidad similar al parénquima renal; 3.) distribución homogénea del material de contraste en el tumor, sin acúmulos ni "shunts" arteriovenosos; y 4.) apariencia en "radios de rueda" de las arterias que lo irrigan. Puede verse, radiológicamente, una zona central hipovascular que corresponde a la zona cicatricial que se ve en el especimen macro.
En algunos casos hay lesiones múltiples: oncocitosis (u oncocitomatosis), y pueden coexistir con carcinomas renales. Un hecho que parece tener muchas implicaciones en su histogénesis es la frecuencia con la que se detectan carcinomas cromófobos en los casos de oncocitosis, demostrando la estrecha relación entre estos dos tumores.
Hallazgos macroscópicos: Son lesiones esféricas bien definidas, usualmente solitarias (salvo en oncocitosis), sin cápsula o con una delgada pseudocápsula de parénquima comprimido que lo rodea; su dimensión es, en promedio 3 - 6 cm, pero pueden mediar más de 12 cm; tienen aspecto homogéneo, color marrón o un tono caoba; se identifica una zona central de aspecto fibroso (o "cicatricial") en una tercera parte de los casos (no específica de oncocitoma); no presentan necrosis ni hemorragia visible macroscópicamente. Pueden extenderse a la grasa perirrenal. En ocasiones son multicéntricos o bilaterales (3-5%).
Hallazgos microscópicos: Está formado exclusiva o predominantemente de células eosinofílicas con citoplasma granular amplio (oncocitos); las células se disponen en nidos o túbulos, hay zonas con aspecto trabecular y áreas compactas, sólidas. Muchos de los grupos celulares, principalmente en las zonas menos periféricas, están embebidos en un estroma poco celular, edematoso o laxo, muchas veces hialinizado; ocasionalmente hay microquístes. Los núcleos son generalmente redondos, bien definidos, con nucléolos visibles. Hay áreas de céluals más pequeñas, con poco citoplasma, núcleos más homogénos y redondos y un agrupamiento celular más denso, las células de estas áreas han sido llamdas oncoblastos. Es muy frecuente y carcaterísticos de oncocitoma encontrar núcleos grandes, irregualres, hipercromáticos, sin nucléolos y con relación núcleo/citoplasma conservada, estos cambios corresponden a cambios degenerativos, no verdadera atipia, similares a los descritos en otras lesiones benignas como el schwanoma. No hay halos perinucleares como en el cromófobo (o estos son ocasionales). Es excepcional encontrar metaplasia ósea [ver imagen (link)]

Figura 20. Células eosinofílícas con citoplasma granular amplio (oncocíticas), que se disponen formando túbulos o nidos, núcleos centrales, relativamente homogéneos y con nucléolos pequeños . No hay, o son escasas, las células con halo perinuclear. (H&E, X400).

Figura 21. En muchos oncocitomas podemos ver áreas con células más pequeñas, mayor relación núcleo/citoplasma y núcleos redondos, homogéneos, con nucléolo inconspicuo. Estas células han sido llamadas "oncoblastos" y son carcaterísticas de este tumor, aunque también pueden verse células con aspecto similar en otros tumores de células granulares. (H&E, X400).
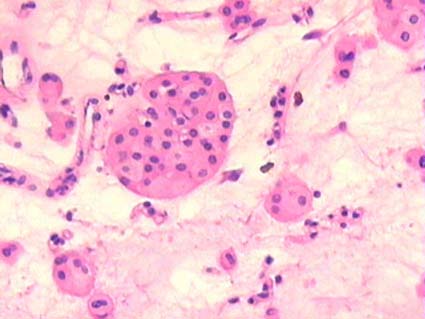
Figura 22. Es muy frecuente encontrar, en oncocitomas, áreas centrales con degeneración mixoide del estroma, en medio de él se ven, "sueltas", células aisladas y en pequieños grupos. (H&E, X400).

Figura 23. Un razgo carcaterístico de oncocitomas son los cambios nucleares degenerativos que se ven en algunos casos. Suelen ser focales, sin mitosis y con relación núcleo/citoplasma conservada; no se identifican nucléolos. Observe la vacuolización de estos núcleos "pseudoatípicos". Este hallazgo también lo podemos ver, aunque menos frecuente, en carcinomas cromófobos. (H&E, X400).
Es posible (permisible) encontrar en algunos oncocitomas compromiso focal de la grasa perirrenal [imagen (link) y otra más (link)], invasión microvascular, necrosis focal, hemorragia, ocasionales mitosis no atípicas y ocasionales papilas. Pero, es incompatible con el diagnóstico de oncocitoma la presencia de un componente de algún tipo de carcinoma, necrosis extensa, áreas fusiformes o sarcomatoides, abundantes mitosis o mitosis atípicas, compromiso de la vena renal, presencia de grupos de células claras (puede haber algunas aisladas) y arquitectura papilar prominente. Algunos autores consideran que si hay más de una mitosis por 20 campos de gran aumento debe clasificarse como un carcinoma cromófobo.
[Varias imágenes microscópicas de muy buena calidad (link)]
Por microscopía electrónica el citoplasma muestra abundantes mictocondrias que lo llenan, con aspecto edematizado y la aparente disminución de otras organelas. La tinción con el hierro coloidal de Hale es negativa o hay una tinción focal en el citoplasma, carcaterísticamente apical; un hallazgo bastante útil en el diagnóstico diferencial con carcinoma cromófobo.

Figura 24. La tinción con el hierro coloidal de Hale es negativa o con marcación del borde citoplasmático apical en oncocitomas (flechas); un aspecto muy diferente al de tinción fuerte, difusa, en carcinomas cromófobos. Compare con la misma tinción en un CCR cromófobo en la Parte 1. (H&E, X400).
Por IHQ hay positividad para CK8 y CK18 (a veces con un carcaterístico patrón punteado), CK14, CD3, parvalbumina y para el protooncogen RON (también el carcinoma cromófobo). Son negativos para CK7, CK20, vimentina., RCC, CD10 y E-cadherina. La positividad para CK7 puede ser débil y focal, pero no intensa y difusa como en el cromófobo.
Citogenética: Es variable, se decriben pérdidas en los cromosomas 1, 14 e Y (también descritas en carcinoma cromófobo); t(5;11) y t(11q;9p) y cambios en el ADN mitocondrial. No hay alteraciones en 3p ni las polisomías características del carcinoma papilar.
Diagnóstico diferencial: debe diferenciarse del carcinoma convencional con células granulares: usualmente encontramos áreas de células claras, hay más mitosis, mayor variabilidad nuclear y, en lesiones con estadio avanzado, invasión extensa perirrenal, necrosis y extensa hemorragia. El diagnóstico diferencial más problemático es con el carcinoma cromófobo; estos últimos, contrario a los oncocitomas, tienen halos perinucleares (no siempre), en un extenso muestreo encontraremos, en muchos casos, áreas carcaterísticas de cromófobo, presentan positividad intensa y difusa para hierro coloidal a pH <1,9; hay positividad para CK7 intensa y difusa en la mayoría de casos (negativa o débil y focal en oncocitomas) (ver Tabla 5).
Tumores metanéfricos
Son tumores benignos poco frecuentes (0,2% de neoplasias epiteliales en riñones de adultos), con histogénesis incierta, más comunes en la edad media: 38-64 años y más frecuentes en mujeres: relación hombre:mujer 1:2. También han sido llamados "tumor renal metanefroide", "adenoma similar a nefroblastoma" y "nefroma nefrogénico". Los pacientes son asintomáticas y suele detectarse como un hallazgo incidental. En algunos casos hay policitemia (al parecer como un fenómeno paraneoplásico), hematuria o masa en el flanco. Macroscópicamente son lesiones sólidas, bien circunscritas, a veces lobuladas, de color grisáceo y tamaño variable, entre 0,5 y 15 cm.
Algunos autores han postulado que pueden representar formas maduras del tumor de Wilms ya que recuerdan restos nefrogénicos y formas maduras del tumor, tanto por histología como por IHQ (Muir T, et al., Metanephric adenoma, nephrogenic rests, and Wilms' tumor: a histologic and immunophenotypic comparison. Am J Surg Pathol 25:1290-6, 2001 [PubMed link]), aunque el 56% tienen cambios en 2p13, una alteración no encontrada en el tumor de Wilms. También se ha relacionado con lesiones papilares benignas (Pesti T, et al., Mapping a tumor suppressor gene to chromosome 2p13 in metanephric adenoma by microsatellite allelotyping. Hum Path 32:101-4, 2001)[PubMed link]).
Hallazgos microscópicos: Histológicamente recuerdan el epitelio tubular metanéfrico. Están formados por células pequeñas, homogéneas, con escaso citoplasma y núcleos redondos, hipercromáticos, sin nucléolos visibles o inconspicuos y que se disponen formando túbulos y acinos pequeños, uniformes, que se mezclan con áreas sólidas. El estroma es mínimo y acelular, pero puede ser edematoso y/o hialinizado. Pueden haber pequeñas papilas que recuerdan cuerpos glomeruloides del nefroblastoma. Es frecuente encontrar microcalcificaciones y cuerpos de psammoma; es más inusual la presencia de necrosis y hemorragia. Hay pocas o ninguna mitosis. [7 muy buenas imágenes microscópicas (link)]

Figura 25. Adenoma metanéfrico: observe el tipo de células, con poco citoplasma y nucléolos inconspicuos. Se disponen en microtúbulos y áreas de aspecto sólido. El estroma es escaso y acelular. Algunas áreas pueden ser muy similares a carcinomas papilares conpatrón tubular. No suelen verse mitosis o son muy escasas. En el adenofibroma metanéfrico hay áreas similares a estas que se mezclan con zonas fusocelulares sin atipia. (H&E, X400).
Por histoquímica no se detecta glucógeno ni mucinas. Ultraestructuralmente se detectan cilios en la superficie luminal de muchas de las células y gránulos apicales secretorios. Por IHQ hay expresión de citoqueratinas, peanut lecitina, CD57, vimentina y WT-1; la positividad para CK7 es focal. Hay negatividad para EMA. Por citogenética algunos casos presentan alteraciones similares al carcinoma papilar: ganancia del cromosoma 7 y 17 y pérdida del Y.
En el diagnóstico diferencial es muy importante no confundirlos con nefroblastoma del adulto (tumor de Wilms), estos suelen tener más proliferación en el componente blastemal; en el adenoma metanéfrico no hay tal actividad proliferativa y no encontraremos las áreas mesenquimales que se ven en muchos tumores de Wilms. Para diferenciarlo de carcinomas papilares debemos observar bien los núcleos: usualmente hay nucléolos y más alto grado nuclear en estos últimos; además, hay positividad fuerte y difusa para CK7 y EMA y negatividad para CD57 y WT-1 en carcinomas papilares.
Ver Caso 126 de nuestra serie de casos: Adenoma metanéfrico.
Es una lesión benigna en la que se mezcla un componente epitelial similar al del adenoma metanéfrico, con un componente estromal maduro, sin anaplasia ni (o con escasa) actividad mitótica. Anteriormente se le denominaba adenofibroma nefrogénico. Hay poco más de 50 casos reportados en la literatura. Son lesiones de niños y adultos jóvenes (3,5-23 años). Se debe hacer el diagnóstico diferencial con el nefroma mesoblástico congénito. Los adenofibroma metanéfricos son lesiones mal definidas y no encapsuladas.
Pueden presentarse con hipertensión, policitemia y hematuria. Algunos autores creen que surgen de restos nefrogénicos que maduran y se diferencian a elementos mesenquimales.
Hallazgos microscópicos: Hay fascículos de células fusiformes sin atipia nuclear ni mitosis que rodea nódulos de tejido epitelial de aspecto embrionario; este componente epitelial consiste de células cúbicas o columnares pequeñas con núcleos oscuros, sin nucléolos notorios, escaso citoplasma y que se disponen en láminas, túbulos o papilas, algunas veces formando estructuras glomeruloides. Los márgenes de la lesión son irregularmente infiltrativos y engloban túbulos y glomérulos adyacentes. Es frecuente encontrar cuerpos de psammoma.
Diagnóstico diferencial: Con nefroblastoma: no hay, en el adenofibroma metanéfrico, el indice de proliferación y actividad mitótica del nefroblastoma, además no es un tumor encapsulado y su estroma interdigita con las estructuras del parénquima renal (contrario a lo que suele verse en el nefroblastoma). Con el nefroma mesoblástico congénito: Las células mesenquimales pueden ser muy similares y presentar el mismo patrón infiltrativo, pero las células del adenofibroma no muestran positividad para actina o desmina ni presentan las características miofibroblásticas del nefroma mesobástico.
Hay informes de algunos casos de lesiones que mezclan características de adenoma metanéfrico con un componente estromal maligno, positivo para vimentina y CD34 y negativo para marcadores epiteliales, lo han denominado adenosarcoma metanéfrico, pero, no aparece en la clasificación de tumores renales de la OMS (Tabla1).
Es una neoplasia renal pediátrica de reciente descripción. Se presenta como una lesión fibrosa centrada en la médula renal, que contiene quistes de pared delgada. Microscópicamente es idéntico al componente estromal del adenofibroma metanéfrico; las células neoplásicas son fusiformes y se disponen en forma arremolinada (en "bulbo de cebolla") alrededor de túbulos atrapados; puede haber diferenciación heteróloga en forma de cartílago, glia o varios tipos de alteraciones vasculares. Por IHQ muchos casos son positivos para CD34. Es una lesión benigna y su tratamiento es quirúrgico.
Tumores mesenquimales
Entre los tumores mesenquimales de adultos la mayoría tienen aspectos morfológicos y clínicos similares a los de otras localizaciones (principalmente tejidos blandos), pero, hay tres que son carcaterísticos del riñón: angiomiolipoma, tumor de células yuxtaglomerulares y tumor renomedular de céluals intersticiales.
Son lesiones benignas formadas por vasos de pared gruesa y cantidades variables de tejido adiposo y músculo liso. Aunque su histogénesis no es completamente clara, se cree que se originan de células epitelioides perivasculares con el potencial de diferenciación hacia estos tres tejidos (PEComas "Perivascular Epitheliod Cells"). Es una lesión monoclonal, por lo tanto no es un hamartoma o un coristoma, sino, una verdadera neoplasia.
Corresponde a menos del 1% de neoplasias renales y se presenta en adultos. Muchos casos son esporádicos (60-70%), pero los restantes se relacionan con alteraciones genéticas como la esclerosis tuberosa en la que hay alteraciones en el gene TSC1 (9q34, que codifica la hamartina) y en el gene TSC2 (16p13.3, que codifica la tuberina, que interactúa con la hamartina); en esta enfermedad hay tumores del SNC, retina, piel (angiofibromas), corazón (rabdomiomas), hueso, pulmón (linfangioleiomiomatosis), riñón (angiomiolipomas y carcinomas); puede haber también retardo mental y convulsiones desde la infancia. Es una enfermedad autosómica dominante.
La edad media de presentación es a los 45 años, pero puede presentarse en niños; la relación hombre: mujer es de 1:4. El diagnóstico puede hacerse con relativa certeza, en muchos de los casos, con los estudios de imágen, ya que presenta aspectos tomográficos muy característicos. Muchos de los casos son asintomáticos, pero en otros puede haber dolor en el flanco, hemorragia masiva por ruptura y falla renal por compresión del parénquima. La frecuencia más alta en mujeres sugiere la posibilidad que algunas hormonas tengan influencia en el crecimiento tumoral.
Hallazgos macroscópicos: Son lesiones usualmente bien definidas y no encapsuladas. El tamaño es muy variable, desde milímetros hasta más de 20 cm; pueden reemplazar extensamente el órgano y, algunas veces, dejar atrapadas estructuras renales en las que podemos encontrar túbulos que se dilatan y le dan un aspecto multiquístico. Suelen ser solitarios, pero pueden ser múltiples (hasta 33%) y bilaterales (15%), más frecuentemente en esclerosis tuberosa. Son de color amarilloso con áreas blanquecina y grisáceas, y tienen apariencia de tejido adiposo, al menos focalmente; los tumores con más contenido de músculo liso son más firmes y pueden recordar leiomiomas. Pueden haber zonas de hemorragia y cambios degenerativos que le dan un aspecto moteado y friable. Pueden invadir ganglios linfáticos regionales y la vena renal, sin que esto signifique malignidad; también es frecuente encontrar invasión capsular. [Imagen macro (link)]
Hallazgos microscópicos: Hay un patrón trifásico de células fusiformes: músculo liso con aspecto relativamente maduro pero dispuesto de una manera desordenada, islas de tejido adiposo también maduro y vasos sanguíneos anormalmente formados, con pared gruesa, muchas veces tortuosos, sin lámina elástica interna o que aparece fragmentada, y con áreas de adelgazamiento y engrosamiento de su pared. En el tejido adiposo podemos ver áreas de necrosis, con lipófagos y células gigantes. El músculo liso se mezcla con la parte externa de los vasos sanguíneos y sus núcleos suelen disponerse perpendiculares a la pared de estos vasos; pueden tener núcleos grandes e irregulares, con hipercromatismo y mitosis y, a veces, adoptar un patrón epitelioide. [Imagen micro / Otra imagen micro / Una más (link)]
Algunos casos aparecen con muy poco o nada de componente adiposo y otros casos están compuestos casi completamnte por tejido adiposo. Los angiomiolipomas compuestos predominantemente (95% o más) por un único componente (tejido adiposo o músculo liso) son llamados monofásicos. Si 95% o o más del tumor es músculo liso es llamado AML "pobre en grasa" (Lane BR, et al. Clinical correlates of renal angiomyolipoma subtypes in 209 patients: classic, fat poor, tuberous sclerosis associated and epithelioid. J Urol. 2008;180:836-43. [PubMed link]). En estos casos la IHQ, demostrando positividad para HMB45 o para melan-A, es esencial para confirmar el diagnóstico.
Ver Caso 67 de nuestra Serie de Casos: Angiomiolipoma pobre en grasa.
Por IHQ hay positividad para marcadores melanocíticos: HMB45, HMB40 y melan-A, al igual que en otros PEComas; las células musculares son positivas para actina y desmina; también encontraremos positividad para CD63 y tyrosinasa. Hay expresión variable de CD68, NSE, S-100 y de receptores estrogénicos y progestágenos (20-28%). Los marcadores epiteliales son negativos.
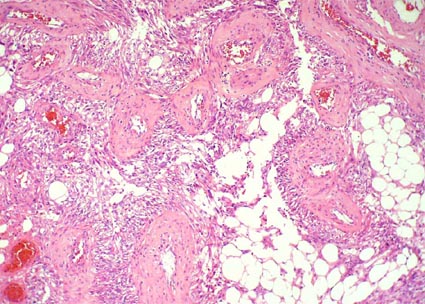
Figura 26. Haces de músculo liso en medio de tejido adiposo maduro y vasos sanguíneos de pared gruesa que se mezclan con los elemntos de tejido muscular y adiposo. Hay positividad para HMB45 en células musculares, adiposas y de vasos sanguíneos de pared gruesa. (H&E, X200).

Figura 26b. Áreas de adipocitos y de células musculares lisas. (H&E, X400).
Por microscopía electrónica detectamos premelanosomas y características de músculo liso y de tejido adiposo.
Citogenética: Alteraciones en los genes relacionados con la esclerosis tuberosa (TSC1 y TSC2 - 9q34 y 6p13.3 respectivamente).
Diagnóstico diferencial: Es posible que en algunas lesiones predominantemente musculares nos planteemos el diagnóstico de carcinoma renal sarcomatoide, leiomioma o leiomiosarcoma; un muestreo extenso de la lesión permitirá descubrir áreas con el característico patrón trifásico; la IHQ es de gran ayuda: positividad para HMB45 u otros marcadores melanocíticos y negatividad para marcadores epiteliales (CK y EMA principalmente).
En el tratamiento de estos pacientes es importante el tamaño de la lesión y el compromiso de estructuras adyacentes. Muchos casos se manejan sin resección, con seguimiento clínico. Hay casos excepcionales informados en la literatura de "transformación sarcomatosa" de angiomiolipoma.
Angiomiolipoma epitelioide
También llamado angiomiolipoma atípico u oncocitoide. Tiene características morfológicas intermedias con carcinoma. Su mayor importancia radica en que pueden tener un comportamiento local más agresivo y pueden, aunque es raro, presentar metástasis.
Hallazgos microscópicos: Hay un extenso componente de células de músculo liso poligonales con citoplasma eosinofílico abundante y una apariencia epitelioide parecida a la de oncocitomas; su tamaño es variable al igual que su aspecto atípico, desde mínimo hasta marcados cambios atípicos incluyendo células gigantes multinucleadas. Podemos ver áreas fusiformes y células mononucleadas grandes con núcleos excéntricos y nucléolos prominentes que recuerdan células ganglionares. Pueden ser prominentes la actividad mitótica, necrosis y hemorragia (muchos casos con poca actividad mitótica). El componente de tejido adiposo y vasos de pared gruesa puede ser muy escaso a ausente. En algunos casos hay áreas hialinas, usualmente en banda, que podrían corresponder a vasos esclerosados. Por IHQ hay la misma expresión de moléculas relacionadas con melanosomas (IHQ similar al angiomiolipoma no epitelioide).
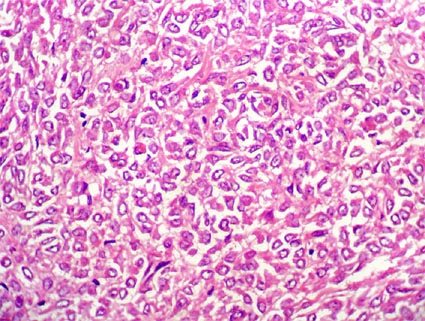
Figura 26c. Angiomiolipoma epiteliode. Células poligonales, algunas con citoplasma amplio, que pueden hacer pensar en tumores epiteliales. Es indispensable un amplio muestreo de la lesión para detectar áreas fusocelulares, vasos de pared gruesa y/o adipocitos. La inmunohistoquímica es de mucha utilidad en casos difíciles (H&E, X400)
Tumor renomedular de células intersticiales (fibroma medular)
También llamado hamartoma medular. Es una lesión común, medular, que forma pequeños nódulos asintomásticos que suelen detectarse incidentalmente en nefrectomías o autopsias. No se sabe con certeza si son verdaderas neoplasias o lesiones hiperplásicas secundarias a hipertensión. Suelen detectarse en pacientes mayores de 20 años y son relativamente frecuentes (hasta en casi la mitad de pacientes mayores). En pocas ocasiones protruyen hacia las cavidades pielocaliciales. Las células intersticiales medulares pueden producir prostaglandinas y regular la presión sanguínea intrarrenal, pero no sabemos con certeza si este tumor tiene implicaciones funcionales.
Macroscópicamente son nódulos bien definidos, esféricos, grisáceos, pálidos, de consistencia firme y generalmente localizados en la porción media de la médula, miden 2-5 mm. Microscópicamente son poco celulares, no encapsulados, compuestos de células fusiformes o estrelladas en medio de un estroma similar al de la médula renal; podemos ver bandas de tejido fibroso laxo y, algunas veces, material amiloide. Con frecuencia hay túbulos medulares atrapados en la peroferia de la lesión. [Imagen micro (link)] [Imagen micro (link)]
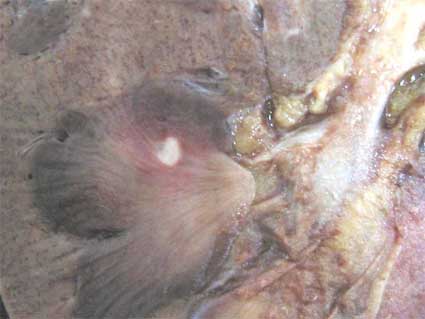
Figura 27. Hallazgo incidental en una autopsia de una mujer de 53 años fallecida por tuberculosis diseminada. Este nódulo de 0,5 cm, blanquecino, en médula, es característico del fibroma medular. Ver el aspecto microscópico en la Figura 27.

Figura 28. Lesión poco celular, con células de aspecto fibroblástico, sin atipia, en medio de un estroma fibrilar y, en muchos casos, con bandas de tejido hialino, eosinofílico como puede verse en ambas imágenes. No suelen identificarse mitosis. La lesión es bien definida pero no encapsulada. (H&E, izquierda, X200; derecha, X400).
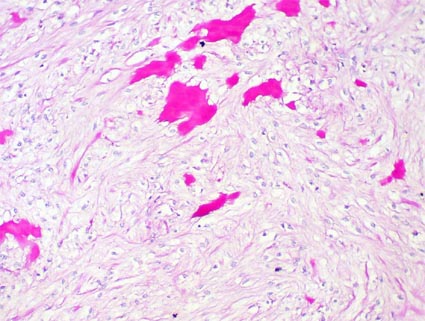
Figura 28b. Fibroma medular. Con la tinción de PAS se resalta algunos acúmulos de material amorfo, muy característico en estas lesiones (PAS, X200).
Principales fuentes bibliográficas:
Bibliografía