
Glomerulonefritis membranosa
La glomerulonefritis membranosa (GNM) es una enfermedad caracterizada por depósitos inmunes subepiteliales, con engrosamiento, usualmente difuso, de las paredes capilares glomerulares y, en muchos casos, la formación de proyecciones perpendiculares de material similar a la membrana basal glomerular (MBG) en la parte externa de ésta (entre el citoplasma del podocito y la MBG): "spikes". Debido a que en esta glomerulopatía no suelen detectarse células inflamatorias y a que en algunos, o muchos de los casos, podría no haber una prominente inflamación local, sino, atrapamiento de complejos imunes, algunos autores prefieren utilizar el nombre glomerulopatía membranosa o glomerulonefropatía membranosa, sin embargo, la presencia de inmunoglobulinas, complemento y complejo de ataque de membrana (C5b-9) implica un proceso inflamatorio (ver más adelante). Otros términos utilizados han sido: nefropatía membranosa y nefropatía (o glomerulopatía) epimembranosa, perimembranosa o extramembranosa.
La GNM es más comunmente una enfermedad primaria o idiopática, pero también se presenta como una enfermedad secundaria a otras condiciones, principalmente infecciones, neoplasias y lupus eritematoso sistémico (LES). Aproximadamente una cuarta parte de casos son secundarios, siendo mayor el porcentaje en niños y ancianos (Glassock RJ, Nephrol Dial Transplant 7(S):64-71, 1992 [PubMed link]). Los hallazgos histopatológicos no permiten una diferenciación entre formas primarias o secundarias, sin embargo, algunas características microscópicas y hallazgos inmunopatológicos (depósitos de complemento que indiquen activación de la vía clásica: C1q, C4) hacen sospechar formas secundarias.
La GNM es la causa más común de síndrome nefrótico en adultos caucásicos (la glomeruloesclerosis focal y segmentaria es la más común en afroamericanos e hispánicos (Arias LF, et al. Glomerular diseases in a Hispanic population: review of a regional renal biopsy database. Sao Paulo Med J. 2009;127(3):140-4. [PubMed link][Free full text]) Mejía G, et al. Descripción clínico-patológica de las enfermedades glomerulares. Estudio de 383 biopsias renales. Acta Médica Colombiana 14: 369-374, 1989). Esta enfermedad responde por aproximadamente el 21-35% de casos de síndrome nefrótico en adultos y 1,5-9% en niños. Muchas series muestran mayor frecuencia de GNM en hombres, con una relación H:M de 2:1.
La presencia de inmunoglobulinas (Igs) y fracciones del complemento en las paredes capilares, y las similitudes morfológicas e inmunopatológicas entre la GNM y enfermedades glomerulares inmunológicas experimentales, respaldan el concepto que la GNM es una enfermedad mediada por complejos inmunes. Los depósitos provienen de complejos inmunes circulantes en algunos casos, pero en la mayoría corresponden a formación in situ por anticuerpos circulantes que reconocen antígenos nativos del glomérulo o antígenos extraños a él que se han depositado allí.
La GNM es probablemente una enfermedad heterogénea en la que un común denominador podría ser que los podocitos proveen el blanco antigénico para la formación in situ de depósitos inmunes glomerulares.
Desde hace muchos años se conocen la similitudes morfológicas entre la GNM y la nefritis experimental de Heymann. En ésta, se inmunizan ratas contra antígenos de la corteza renal; los animales desarrollan una enfermedad clínica y morfológicamente similar a la GNM.
Estudios iniciales en este modelo sugerían que los depósitos resultaban de atrapamiento de complejos inmunes circulantes formados por antígenos del borde en cepillo de túbulos con su correspondiente anticuepo. Esta hipótesis se basaba en la observación que la enfermedad se inducída por fracciones de preparados del borde en cepillo, no de extractos glomerulares. Subsecuentemente, el desarrollo del modelo de nefritis pasiva de Heymann en ratas que recibían una inyección de anticuerpos anti-borde en cepillo llevó a sugerir que los depósitos inmunes subepiteliales podrían formarse sin la intervención de complejos inmunes circulantes. Otros autores demostraron que los anticuerpos anti-borde en cepillo podían unirse al glomérulo en la ausencia de antígenos circulantes del borde en cepillo, lo cual probó que la formación de complejos inmunes ocurría in situ. La evidencia definitiva que estableció el papel de la formación in situ de estos complejos en la pared capilar glomerular requiró la identificación del antígeno. Como dato histórico, un blanco del anticuepo en ratas fue identificado por Kerjaschki y Farquhar (Kerjaschki D, Farquhar MG: Proc Natl Acad Sci U S A 79 : 5557 –5561, 1982 [PubMed link] [Free Full Text] / Kerjaschki D, Farquhar MG. J Exp Med 157 : 667 –686, 1983 [PubMed link][Free Full text]) a principios de los 80 como una proteína de la membrana del podocito ahora llamada megalina. El receptor megalina, un miembro de la superfamilia de receptores LDL, se expresa en el extremo del proceso podocitario (donde se forman los complejos inmunes) (Ronco P, Debiec H. Molecular pathomechanisms of membranous nephropathy: from Heymann nephritis to alloimmunization. J Am Soc Nephrol. 2005;16(5):1205-13. [PubMed link][Full Text link])
Los antígenos identificados en la nefritis de Heymann son antígenos del borde en cepillo de los túbulos renales; o una glicoproteína, megalina, sintetizada por las células epiteliales viscerales del glomérulo. En la GNM humana se ha logrado demostrar presencia de anticuerpos contra antígenos del borde en cepillo en muy pocos casos. En las años setenta, un grupo japonés demostró localización de antígenos tubulares en los depósitos inmunes de pacientes con GNM (Naruse T et al, J Exp Med 144:1347-62, 1976 [PubMed link] [Free full text]), sin embargo, grupos de otros centros no han logrado los mismos hallazgos (Collins AB, et al, Nephron 27:297-301, 1981 [PubMed link]; Thorpe LW y Cavallo T, J Clin Lab Immunol 3:125-127, 1980 [PubMed link]; Whitworth JA, et al, Clin Nephrol 5:159-162, 1976 [PubMed link]). En el momento actual la evidencia sugiere que el complejo de antígenos-anticuerpos (Ags-Acs) de la nefritis de Heymann tiene poco que ver en la GNM. La variedad de Ags-Acs asociados con formas secundarias de GNM sugiere que en las formas idiopáticas la presentación morfológica es común a múltiples complejos Ags-Acs.
La endopeptidasa neutra (un antígeno del podocito que puede digerir biológicamente peptidos activos) fue identificada como el blanco antigénico de anticuerpos depositados en el espacio subepitelial de los glomérulos en un grupo de pacientes con GNM prenatal. Las madres se hacen inmunizadas porque tienen deficiencia de endopeptidasa neutra debido a mutaciones en su gen (Ronco P, Debiec H. New insights into the pathogenesis of membranous glomerulonephritis. Curr Opin Nephrol Hypertens. 2006;15:258-63. [PubMed link]).
Más recientemente (en un trabajo publicado en julio de 2009), un grupo de investigadores, usando western blot de extractos de proteínas de glomérulos humanos normales con muestras de suero de pacientes con glomerulopatía membranosa, encontraron que la mayoría de pacientes con nefropatía membranosa idiopática tienen anticuerpos contra un epítope en el receptor de la fosfolipasa A2 de tipo M (PLA2R), indicando que PLA2R es un antígeno importante en esta enfermedad (Beck LH, et al. M-Type Phospholipase A2 Receptor as Target Antigen in Idiopathic Membranous Nephropathy. N Engl J Med. 2009;361(1):11-21. [Extract link]). En un trabajo reciente (2010) se informa co-localización de anticuerpos específicos anti-aldosa reductasa (AR) y anti-manganeso superoxido dismutasa (SOD2) con IgG4 y C5b-9 en los depósitos electon-densos de podocitos. Estos datos sugieren que AR y SOD2 son antígenos renales en la GN membranosa y que el estrés oxidativo puede inducir expresión de SOD2 glomerular (Prunotto M, et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol. 2010;21(3):507-19. [PubMed link]).
La endopeptidasa neutra y PLA2R son dos antígenos que, en condiciones no patológicas, se localizan en la membrana del podocito. Sin embargo, los niveles de anticuerpos anti-endopeptidasa neutra en adultos con GNM no han sido diferentes de controles aparentemente saludables, sugiriendo que no están involucrados en GNM primaria, por lo que su determinación en pacientes con GNM parece de poca utilidad. Los datos para especificidad de anticuerpos anti-PLA2R parecen sólidos y su valoración podría ser muy útil para clínicos, ayudando a diferenciar GNM primaria de secundaria (Murtas C, et al. Circulating antipodocyte antibodies in membranous nephropathy: new findings. Am J Kidney Dis. 2013;62(1):12-5. [PubMed link]).
En 2014, Tomas NM et al publicaron que aproximadamente 2,5 a 5% de los pacientes con GNM idiopática evaluados tenían autoanticuerpos contra "thrombospondin type-1 domain-containing 7A" (THSD7A), lo cual corresponde a 8 a 14% de los pacientes seronegativos para anti-PLA2R1. THSD7A fue inicialmente considerado una proteína endotelial que se expresa en la vasculatura placentaria. Los autores encontraron que THSD7A se concentra en la parte basal del podocito y colocaliza con nephrina en la inmunofluorescencia, y ellos no encontraron expresión en las células endotelales glomerulares. (Tomas NM, Beck LH, 0Meyer-Schwesinger C, et al. Thrombospondin Type-1 Domain-Containing 7A in Idiopathic Membranous Nephropathy. N Engl J Med 2014; 371:2277-87 [Article on NEJM - link]; Alsharhan L, Beck LH Jr. Membranous Nephropathy: Core Curriculum 2021. Am J Kidney Dis. 2021 Mar;77(3):440-453. [PubMed link] [Free full text]; Sethi S. New 'Antigens' in Membranous Nephropathy. J Am Soc Nephrol. 2021 Feb;32(2):268-278. [PubMed link]).
Al menos 14 blancos antigénicos han sido identificados antes de 2023, que representan el 80-90% de casos de GNM: PLA2R, THSD7A, NELL1, SEMA3B, PCDH7, HTRA1, NTNG1, EXT1/EXT2, NCAM1, TGFBR3, CNTN1, FAT1, NDNF, PCSK6. Muchas de las formas de GNM asociadas con estos nuevos antígenos tienen fenotipos clínicos y patológicos distintivos. En 2023, se describieron 17 supuestos antígenos adicionales, lo que redujo significativamente nuestra brecha de conocimiento sobre los blancos antigénicos de la enfermedad. Estos incluyen "seizure-related 6 homolog-like 2" (SEZ6L2), vasorina (VASN), antígeno endosómico temprano 1 (EEA1), estimulante de macrófagos 1 (MST1), receptor del péptido natriurético 3 (NPR3), ficolina 3 (FCN3), CD206, proteína de neurona motora rica en cisteína (CRIM1), proteína transmembrana repetida rica en leucina (FLRT3), enzima degradante de insulina (IDE), "reversion-inducing cysteine-rich protein with Kazal motifs" (RECK), neuroligina 3 (NLGN3), "peptidoglycan recognition protein 1" (PGLYRP1), factor A de crecimiento endotelial vascular (VEGFA), sulfatasa 1 (SULF1), proteína 2 de matriz extracelular de fibulina que contiene factor de crecimiento epidérmico (EFEMP2) y proteína de matriz síndrome de Fraser 1 (FRAS1). La evidencia sobre la patogenicidad, las asociaciones clínico-patológicas y el papel clínico de los anticuerpos contra cada uno de los antígenos antes mencionados varía en la literatura (Bonilla M, et al. Hope or hype? Clinicians' dilemma in the era of ever-expanding antigens in membranous nephropathy. Nephrol Dial Transplant. 2023 Nov 30;38(12):2666-2669. [PubMed link]).
El Consenso de la Clínica Mayo sobre GNM, publicado en 2023, propone una clasificación en 2 pasos. El primer paso, cuando sea posible, es la identificación del antígeno diana, basándose en un algoritmo de varios pasos y utilizando una combinación de serología, tinción del tejido de la biopsia renal mediante inmunofluorescencia o inmunohistoquímica y/o por medio de espectrometría de masas. El segundo paso es la búsqueda de una posible enfermedad subyacente o afección asociada, lo cual es particularmente relevante cuando se dispone del conocimiento del antígeno diana. El consenso reconoce que es posible que los recursos y el equipo necesarios para realizar las pruebas propuestas no estén disponibles en general. Sin embargo, el consenso de la reunión fue que ha llegado el momento de adoptar una clasificación de GNM basada en antígenos porque este enfoque permitirá un diagnóstico preciso y específico de la glomerulonefritis, con implicaciones significativas para el manejo del paciente y el tratamiento dirigido (Sethi S, Beck LH Jr, Glassock RJ, et al. Mayo Clinic consensus report on membranous nephropathy: proposal for a novel classification. Kidney Int. 2023 Dec;104(6):1092-1102. [PubMed link]).
Se cree que en humanos es necesaria la activación del complemento para que se produzca el síndrome nefrótico, con formación del complejo de ataque de membrana (MAC) (Groggel GC, et al, J Clin Invest 72:1948-1957, 1983 [PubMed link] [Free full text]), lo que soporta un papel citolítico del complemento.
GNM y hepatitis B: La forma más frecuente de glomerulopatía en pacientes infectados con este virus es la GNM seguida por la GN membranoproliferativa. Los antígenos core (HBcAg) y e (HBeAg) parecen los más importantes en la patogénesis de la GNM. En estos casos se identifican los antígenos, o sus anticuerpos, en los depósitos inmunes glomerulares. No está claro qué se deposita primero: el Ag., el Ac. o el complejo Ag-Ac formado previamente (circulante). La prevalencia de GNM en la infección no se sabe con certeza, pero en niños con GNM se detecta el estado de portador del virus en alrededor de un 20%, con tasas más altas en países endémicos. En adultos la tasa de portador del virus de la hepatitis B en pacientes con GNM es más baja que en niños. En la GNM asociada con esta infección es más frecuente encontrar hipercelularidad mesangial, proliferación endocapilar, depósitos inmunes mesangiales, depósitos inmunes subendoteliales y estructuras tubuloreticulares endoteliales (microscopía electrónica). Es frecuente que se presente con hipocomplementemia. El pronóstico de la GNM en estos casos parece más favorable, con mayor frecuencia de remisión y menos probabilidad de evolución a daño renal terminal.
En hepatitis C también puede presentarse GNM secundaria, aunque es más frecuente en estos casos la GN membranoproliferativa. En muchos estudios no se han logrado identificar antígenos del virus, o Acs contra estos, en los depósitos glomerulares. La clínica puede ser similar a la de la GNM idiopática o pueden presentarse con proteinuria asintomática.
Sifilis congénita: La GNM es una complicación rara, pero bien reconocida causa de síndrome nefrótico en niños con esta infección. Otras complicaciones glomerulares incluyen: síndrome nefrítico agudo y GN proliferativa extracapilar (clínicamente presentándose como una GN rápidamente progresiva). Nosotros hemos visto casos con estos tipos de enfermedad glomerular en quienes hay una mejoría dramática con el tratamiento antibiótico. Varios estudios han demostrado la presencia de antígenos del Treponema pallidum en los depósitos inmunes glomerulares.
En LES la presentación histopatológica es muy variable y hay combinación de alteraciones morfológicas: características de GNM y depósitos imunes subendoteliales, proliferación endocapilar y/o mesangial, semilunas, combinación con características de GN membranoproliferativa, etcétera. En la clasificación histopatológica más reciente de nefrítis lúpica, la GNM pura (clase V) sólo se diagnostica si no hay otras lesiones activas; si se combina con estas otras lesiones se diagnostica como una combinación de clase V y clase III ó IV sólo si hay lesión con características de membranosa en más del 50% del penacho de más del 50% de los glomérulos. Ocasionales depósitos subepiteliales y formación de "spikes" son muy frecuentes en las nefritis lípicas clase III y IV. En la mayoría de estos casos se demuestran depósitos glomerulares de C1q.
GNM y neoplasias: Las neoplasias más frecuentemente asociadas con GNM son carcinomas de pulmón, mama, colon, estómago y riñón, leucemias y linfomas Hodgkin y no Hodgkin; pero, hay informes de GNM en muchos otros tipos de cancer. La incidencia de cancer en pacientes con GNM es de aproximadamente 1%. Los hallazgos histológicos e inmunopatológicos y la presentación clínica son similares a los de las formas idiopáticas de la GNM. La asociación entre la glomerulopatía y la neoplasia está soportada por el curso clínico, la respuesta inmune del huesped al tumor y la patología glomerular, sin embargo, en muy pocos casos se logra documentar un antígeno del tumor, o su anticuerpo, en los depósitos glomerulares. Podría ser que la respuesta inmune contra la neoplasia, en un contexto genético propicio, lleve al desarrollo de la GNM. El pronóstico de la glomerulopatía está supeditado al de la neoplasia. Si hay curación de esta última, la GNM tiende a desaparecer.
Ver el Caso 144 de nuestra Serie de Casos: GN membranosa con depósitos monoclonales de IgG/kappa.
Otras enfermedades asociadas con GNM son: medicamentos y toxinas (oro, penicilamina, mercurio, captopril, etc.), otras infecciones (parásitos, Streptococus), otras enfermedades autoinmunes (artritis reumatoidea, pénfigo, cirrosis biliar primaria, enteropatía autoinmune, enfermedad de Hashimoto, enfermedad de Graves, etc.), diabetes, sarcoidosis, crioglobulinemia, anemia de células falciformes, etc.
Clínica: La presentación más frecuente es proteinuria en rango nefrótico, con o sin los otros hallazgos del síndrome completo. Un porcentaje variable de casos se presenta como proteinuria asintomática. Hay hematuria microscópica en la mayoría de pacientes, pero la hematuria macro es rara. Excepcionalmente puede presentarse como hematuria aislada. La función renal puede estar levemente alterada al momento del diagnóstico en muchos casos, pero la falla renal es poco usual en este momento de la evolución. En una tercera o cuarta parte de los casos se documenta hipertensión arterial sistémica. Puede presentarse a cualquier edad, con predilección por las 4º y 5º décadas de la vida.
El curso clínico de la GNM es muy variable, en muchos pacientes hay una evolución poco agresiva; aproximadamente un 25% de pacientes tendrá una remisión espontánea parcial o completa, aunque, hasta un 29% de ellos presentará reacaídas. Alrededor de un 50% de pacientes no presentará alteración grave de la función renal. En un pequeño número de casos habrá una pérdida rápida de la función o muerte. Esta evolución variable hace difícil la interpretación de resultados clínicos de seguimiento y tratamiento. El tratamiento con esteroides o con clorambucil u otros inmunosupresores ha mostrado resultados contradictorios, no existe en el momento actual un tratamiento universalmente aceptado.
Casos de GNM recurrente post-trasplante han sido informados, pero, no hay series grandes que permitan determinar una incidencia más precisa del porcentaje de recidiva post-trasplante. Dado que los receptores de trasplante renal son suceptibles a muchas causas de GNM secundaria, debe buscarse una causa subyacente o asociada. Histológicamente no es posible diferenciar la GNM recurrente de la GNM de novo en un riñón trasplantado; para esta diferenciación es indispensable el estudio histológico en el riñón nativo (pre-trasplante).
Datos de laboratorio: La proteinuria se encuentra generalmente en rango nefrótico y en muchos casos es severa (>10 g/24h). Se encuentran, además, en la mayoría de casos, los demás hallazgos del síndrome nefrótico completo (hipoalbuminemia, hipercolesterolemia). La proteinuria suele ser no selectiva. Es común la hematuria micro e inusual la hematuria macro. En muchos casos hay elevación leve de creatinina sérica y BUN, y en hasta el 75% de casos hay descenso de la tasa de filtración glomerular. No hay hipocomplementemia (al menos en las formas idiopáticas) y en muchos casos se documentan niveles incrementados del MAC (C5b-C9). Como lo expresamos antes, en un porcentaje alto de casos se detectan complejos inmunes circulantes, aunque, al igual que la detección de nivels incrementados de MAC, su significado en el diagnóstico diferencial es poco, dado que no tienen ninguna especificidad.
Histopatología
Los cambios característicos de la GNM están en las paredes de capilares glomerulares. La fase inicial de la glomerulopatía está marcada por depósitos granulares subepiteliales: en la parte externa de la MBG, entre ésta y el citoplasma de podocitos. Inicialmente estos depósitos no generan reacción de la MBG y por lo tanto es muy poco probable que se detecten con microscopía de luz convencional. En pocos casos los depósitos tienen un tamaño y agregación tal que pueden se visibles, con la tinción de tricrómico, como pequeños granos fuschinofílicos (rojos), algo homogéneamente espaciados, y apoyados en la parte externa de la membrana basal (Figura 3). Para lograr verlos se necesitan cortes histológicos delgados y observar con gran aumento utilizando aceite de inmersión. En cortes tangenciales de la MBG, con tinción de plata-metenamina, puede observarse, en algunos casos, un aspecto moteado o con minúsculos orificios ("holes") que corresponden a las depresiones vistas con microscopio electrónico de barrido, originadas por los depósitos subepiteliales (Figura 4). En esta fase de evolución de las lesiones: ESTADIO I, el diagnóstico puede ser errado si no se dispone de inmunofluorescencia (IF) o de microscopía electrónica (ME); estos depósitos son de origen inmune y serán positivos para IgG y, en la mayoría de casos, para C3, además, son electrón-densos. Sin IF o ME es muy probable que no podamos diagnosticar este estadio y es frecuente, en estos casos, que se diagnostique como cambios glomerulares mínimos.
En el ESTADIO II los hallazgos histológicos en la microscopía de luz convencional permiten un diagnóstico más fácil. La arquitectura glomerular está conservada y las paredes capilares se ven gruesas con las tinciones de rutina (Figura 1). La celularidad no suele estar aumentada (si lo está sugiere una GNM secundaria) y las luces capilares se ven amplias. Hay formación de material con aspecto similar a la MBG (aunque de composición diferente) que se proyecta perpendicularmente a ésta dando el aspecto de puntas o púas ("spikes") (Figura 2). Estos "spikes" se originan en reacción a los depósitos y van progresivamente rodeándolos. Este material está compuesto por colágeno tipo IV, componentes no colagenosos: laminina, proteoglicanos y vitronectina, y podría originarse a partir de estimulos provenientes de mediadores producidos por el podocito o por otro mecanismo que estimule el cambio en la MBG. En algunos cortes se evidencian muy bien los espacios o agujeros ("holes") que producen, en la parte externa de la MBG, los depósitos y el material que los rodea (los "spikes"), con el centro del agujero correspondiendo al depósito inmune. En algunos casos estos "holes" tienen una forma irregular que da un aspecto reticulado a la MBG (Figura 4). En algunos casos evidenciamos proliferación celular segmentaria (Figura 5), pero en estos casos debemos plantearnos la posibilidad de una GNM secundaria; en otros casos habrá también lesiones esclerosantes segmentarias y focales (Figura 6).
Figura 1. En la GNM las alteraciones se evidencian, principalmente, en las paredes capilares; aquí aparecen engrosadas y con aspecto rígido (flechas verdes). En algunos casos encontramos grados variables de hipercelularidad mesangial (flechas azules), en estos casos debemos plantearnos la posibilidad de una causa secundaria. (H&E, X400).

Figura 2. El hallazgo característico y que permite el diagnóstico en la mayoría de casos son las proyecciones perpendiculares en la parte externa de la MBG, evidenciables con la plata (flechas). En estadios más iniciales en los que no se han formado aún los "spikes" puede ser imposible el diagnóstico sin la ayuda de inmunofluorescencia o microscopía electrónica. (Plata-metenamina, X1000).
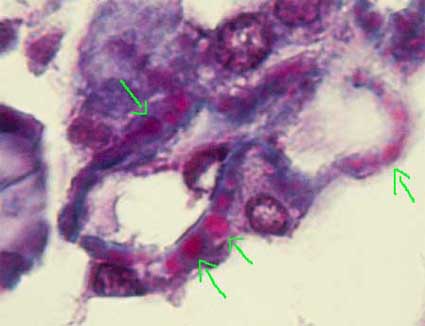
Figura 3. En casos de depósitos subepiteliales grandes es posible, en cortes delgados y con una buena tinción de tricrómico, ver los depósitos inmunes con un característico color rojo (fuschinofílicos) (flechas). Aun en estadios iniciales, sin "spikes", este hallazgo nos permite hacer el diagnóstico de GNM. Desafortunadamente no es frecuente lograr ver estos depósitos con el microscopio de luz convencional (Tricrómico de Masson, X1000).

Figura 4. En cortes en los que la MBG aparece tangencialmente es posible ver espacios como orificios ("holes") en ella (flechas); estos huecos se deben a la presencia de depósitos negativos con la plata rodeados por material de la MBG (positiva con la plata). Muchos de estos espacios se deben a un corte transversal de un depósito rodeado completamente por "spikes". (Plata-metenamina, X1000).

Figura 5. Con la tinción de PAS resalta el engrosamiento de paredes capilares que se ven rígidas; no hay un marcado ensanchamiento mesangial. En este caso hay también un segmento con proliferación endocapilar (flechas); este hallazgo debe hacernos pensar en la posibilidad de una GNM secundaria. (PAS, X400).
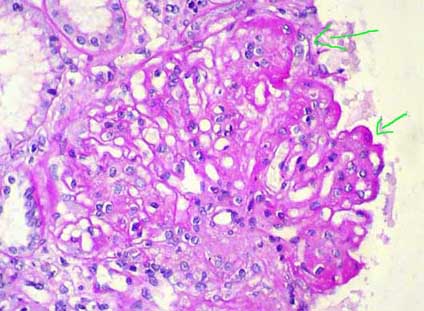
Figura 6. Con alguna frecuencia las alteraciones glomerulares se acompañan de lesiones esclerosantes segmentarias y focales (flechas). Este hallazgo no necesariamente indica un peor pronóstico (PAS, X400).
Al avanzar el proceso el material que forma los "spikes" aumenta y va rodeando completamente los depósitos, se forman así nuevas capas de MBG dejando los depósitas inmersos en esta matriz. Ahora los depósitos se ven intramembranosos y con la tinción de plata pueden tomar un aspecto en "cadena" o en "rosario". En este punto las lesiones se conocen como ESTADIO III. Los depósitos siguen siendo positivos con la inmunotinción, aunque progresivamente se hacen menos electrón-densos (Figuras 7 y 8).

Figura 7. Al avanzar el proceso de la enfermedad los depósitos inmunes parietales van siendo progresivamente rodeados por material similar al de la MBG, lo que le da un aspecto en cadena o "en rosario", irregular a la membrana basal (flechas); estos hallazgos caracterizan al estadio III de la GNM. De acuerdo a las alteraciones predominantes en las paredes capilares diagnosticamos las lesiones como estadio I, II, III, o IV. (Plata-metenamina, X1000).
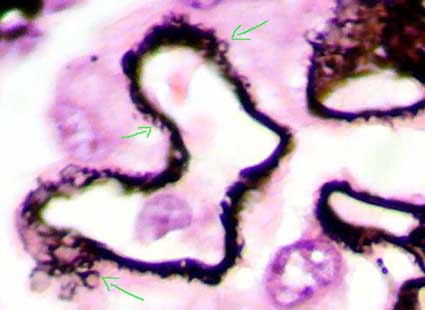
Figura 8. En esta microfotografía se logran evidenciar mejor las lesiones parietales en la GNM estadio III; observe como el material que aparece en la parte externa de la MBG (negro con la plata) forma círculos o anillos que rodean completamente los depósitos inmunes (flechas). (Plata-metenamina, X1000).
En el ESTADIO IV la MBG está irregularmente engrosada, sin la presencia de depósitos electrón-densos o agujeros. En esta fase se considera que los depósitos se han reabsorbido dejando ese engrosamiento irregular. En estos casos el diagnóstico se sustenta por la presencia de otras áreas con lesiones de estadio II o III.
En muchos casos hay una apariencia mixta, con áreas presentando varios estadios. Para clasificar estos casos se requiere una buena observación para determinar el patrón dominante.
Los estadios histopatológicos son progresivos, sin embargo, aunque presentan alguna asociación con la evolución de la enfermedad, no hay una perfecta correlación entre el estadio y el pronóstico. Se informan remisiones en cualquiera de los estadios y progresión a falla renal aun en casos diagnosticados como estadio I y II. Tampoco es claro que estos estadios evolucionen en períodos de tiempo más o menos determinados. En la Figura 9 aparece el esquema de los diferentes estadios de la GNM.

Figura 9. Este esquema representa muy bien las carcaterísticas que definen los diferentes estadios de la GNM. En el estadio I (flechas azul oscuro) los depósitos no han generado aún reacción en la membrana basal y por lo tanto no se acompañan de "spikes". En el estadio II la reacción producida en la parte externa de la MBG ha llevado a la formación de proyecciones perpendiculares a ella ("spikes") que tratan de rodear los depósitos (flechas verdes). En el estadio III el material de la MBG ha rodeado completamente los depósitos (flechas rojas). Y en el estadio IV la MBG esta muy engrosada e irregular y los depósitos han desaparecido casi por completo (flechas azul claro). (Esquema sobre una microfotografía de un corte glomerular teñido con Tricrómico de Masson, X1000).
Otros cambios descritos en GNM son esclerosis segmentaria, lobulación del penacho, hipercelularidad mesangial, presencia de células inflamatorias y necrosis, sin embargo, en estos casos debe sospecharse una forma secundaria. En algunos trabajos se ha documentado coexistencia de GNM y nefropatía IgA, GNM y diabetes, y GNM y GN extracapilar. Ocasionalmente hay casos de GNM con semilunas, en estos casos el curso es severo con mal pronóstico; en varios de estos casos se han detectado anticuerpos anti-MBG.
La formación de "spikes" en la MBG es "casi" diagnóstica de GNM, sin embargo, yo he visto algunos casos en los que el aspecto similar ha llevado a confusiones: amiloidosis con formación extensa de proyecciones perpendiculares a la MBG, glomerulonefritis fibrilar e inmunotactoide y deficit de lecitin colesterol acil transferasa.
El intersticio, túbulos y vasos muestran cambios inespecíficos. Con frecuencia se observan gotas de reabsorción proteica o un aspecto vacuolado del citoplasma de células tubulares. La fibrosis intersticial y atrofia tubular correlacionan con la severidad del daño crónico y son un indicador pronóstico, por lo que deberían cuantificarse o semicuantificarse (leve - moderada - severa). Las causas del daño tubulointersticial, como en muchas glomerulopatías, parecen relacionarse con la alteración de la circulación glomerular y atrofia secundaria. La proteinuria también puede jugar un papel importante en el daño tubular.
Inmunofluorescencia
El cuadro inmunopatológico característico es el de depósitos granulares parietales de IgG acompañados, en aproximadamente el 75% de casos, de C3. La tinción para IgG suele ser más intensa que para C3. La inmunotinción puede verse como gránulos gruesos o como gránulos muy finos y densamente agrupados que le dan un aspecto pseudolineal. Observando detalladamente, puede evidenciarse que estos depósitos están ubicados hacia la parte externa de la MBG. También pueden identificarse, en una minoría de casos, otras inmunoglobulinas, especialmente IgM e IgA. Como fue expresado anteriormente, depósitos de C1q o de C4 obligan a descartar una causa secundaria de GNM; igual sucede si se detectan depósitos mesangiales.
El subgrupo de IgG4 suele ser el más frecuente, este subgrupo fija pobremente el complemento y esto explicaría la tinción más débil para C3 (Doi T et al, Clin Exp Immunol 58:57-62, 1984 [PubMed link]; Nöel LH, et al, Clin Immunol Immunopathol 46:186-194, 1988 [PubMed link]).


Figura 10. La GNM se caracteriza, inmunopatológicamente, por depósitos de IgG y, en la mayoría de casos, C3, granulares en las paredes de capilares. En cortes delgados y a gran aumento es posible, en muchas ocasiones, determinar la localización de los depósitos inmunes en la parte externa de la MBG. Si encontramos también depósitos subendoteliales debemos pensar en la posibilidad de una GNM secundaria. En ambas imágenes se ve un aspecto característico de los depósitos subepiteliales, que a veces describimos como "aspecto reticular". (Inmunofluorescencia con anticuerpos anti-IgG marcados con fluoresceina, X400).
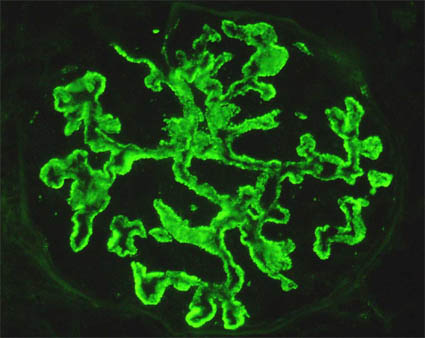

Figura 11. En algunos casos de GNM los depósitos parietales son gránulos que parecen protruir hacia la parte externa de los capilares (como en la foto superior). Otras veces podemos encontrar que los depósitos son muy pequeños y densamente agrupados, que le dan un aspecto pseudolineal (como en la foto infeior). A gran aumento es posible determinar la naturaleza granular de los depósitos para no confundirlos con depósitos lineales que hacen pensar en enfermedad anti-MBG. (Inmunofluorescencia con anticuerpos anti-IgG marcados con fluoresceina, X400).
Microscopía electrónica
Hay depósitos electrón-densos en el lado epitelial (externo) de la MBG, entre ésta y la célula epitelial: Subepiteliales o epimembranosos. Estos depósitos son usualmente difusos y homogéneamente distribuidos, pero pueden encontrarse, en algunos casos, con distribución irregular. Se evidencian los "spikes" como proyecciones irregulares de la MBG entre los depósitos subepiteliales; con la progresión de la enfermedad estas proyecciones se hacen más largas y gruesas y rodean los depósitos incorporándolos en una MBG mucho más gruesa que la original. Los depósitos son amorfos; la presencia de depósitos organizados debe alertar de una posible nefritis lúpica. Estos depósitos van perdiendo su densidad electrónica hasta perderse en los estadíos más avanzados del proceso. Como en muchas otras enfermedades que cursan con síndrome nefrótico, hay una variable pérdida o borramiento de procesos podocitarios. En algunos casos, más frecuentemente secundarios, hay depósitos densos en el mesangio. (Ver imágen de GNM estadio II (link) - Una más de ME (link))

Figura 12a. A la izquierda: depósitos electron-densos en la parte externa de la membrana basal, sin reacción de ésta alrededor de los depósitos: Estadio I. A la derecha: depósitos rodeados lateralmente por material similar al de la membrana basal ("spikes"): Estadio II. Aumento original, X4.000. (Imágenes cortesía del Dr. Carlos A, Jiménez).
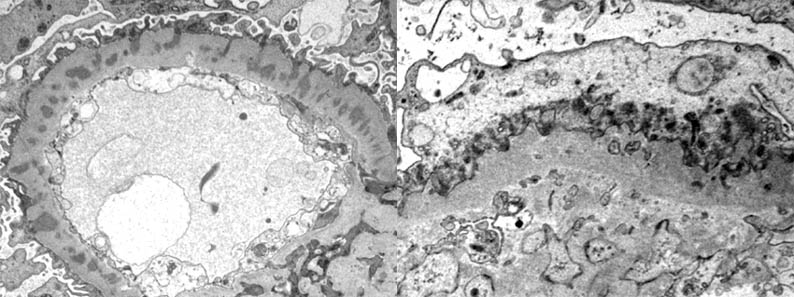
Figura 12b. A la izquierda: los depósitos electron-densos están completamente rodeados por el material similar a la membrana basal, dando el aspecto de estar "metidos" dentro de una membrana basal muy gruesa e irregular: Estadio III. A la derecha: la membrana basal glomerular es gruesa e irregual y los depósitos electron-densos han casi desaparecido: Estadio IV. Aumento original, X4.000 a la izquierda y X6.000 a la derecha. (Imágenes cortesía del Dr. Carlos A, Jiménez). Note la extensa pérdida de los pedicelos en las cuatro imágenes anteriores.
Indicadores pronósticos
Como en la mayoría de glomerulopatías, la creatinina sérica elevada al momento del diagnóstico, proteinuria severa (>10 g/24h), hipertensión arterial, y el daño túbulointersticial crónico se han relacionado, en mayor o menor medida, con un mayor riesgo de evolución a falla renal terminal. Algunos trabajos sugieren mejor pronóstico si la proteinuria es selectiva.
En las formas secundarias hay, en general, un mejor pronóstico si la causa asociada es suceptible de tratamiento. En niños también parece haber un mejor pronóstico. En algunas series se informa mejor pronóstico para mujeres. También los estadios histopatológicos muestran correlación con la evolución, aunque esta correlación no es perfecta como vimos anteriormente.
Bibliografía reciente