
Glomeruloesclerosis focal y segmentaria
La glomeruloesclerosis focal y segmentaria (GEFyS) es una enfermedad caracterizada morfológicamente por segmentos de esclerosis en algunos glomérulos. Puede ser primaria o secundaria y se asocia con síndrome nefrótico, en algún momento de la evolución de la enfermedad, en la mayoría de los casos. Los glomérulos sin lesiones pueden presentar alteraciones de la celularidad mesangial y/o fusión de los procesos podocitarios.
El término esclerosis significa cicatrización y se caracteriza por acumulación de colágeno glomerular (tipo IV). Sin embargo, en la GEFyS algunas de las lesiones glomerulares segmentarias no son necesariamente esclerosis, sino depósitos de material hialino: Hialinosis. De aquí la denominación ya clásica, más de la escuela francesa, de hialinosis focal y segmentaria. Otros investigadores designan a los casos en los que hay segmentos hialinos como: "GEFyS con hialinosis". No hay una explicación clara del porqué en algunos casos hay segmentos hialinos y en otros no. Algunos autores consideran que las lesiones hialinas son precursoras de las lesiones esclerosantes, sin embargo, parece que en muchos casos la lesión esclerosante comienza así desde el principio, sin una fase de hialinosis.
La GEFyS es una enfermedad bien definida clinicopatológicamente. En algunas biopsias renales podemos evidenciar lesiones esclerosantes segmentarias y focales que pueden ser secundarias a episodios anteriores de glomerulonefritis focales; estos casos deben diferenciarse de una verdadera GEFyS, aunque en muchos casos no es una tarea fácil.
Las lesiones histológicas de la GEFyS no permiten diferenciar las formas primarias de las secundarias. Para hacer esta diferenciación debemos ayudarnos de los hallazgos clínicos y de laboratorio y de las alteraciones en otros compartimientos histológicos del tejido (vasos, interstico y túbulos). Aun así no podremos hacer, en muchos casos, una clara distinción entre formas primarias y secundarias. En la medida en que nuestro conocimiento de la etiología y fisiopatogenia de la enfermedad avance podremos distinguir mejor las diferentes formas de la enfermedad. Es posible que en un futuro podamos diagnosticar muchos de estos casos como enfermedades diferentes entre sí de acuerdo a su etiología, y quizá el término GEFyS se torne obsoleto (Cameron JS, The enigma of focal segmental glomerulosclerosis. Kidney Int, Suppl 57, S119, 1996 [PubMed link]).
Hay variantes morfológicas de la GEFyS y para el diagnóstico histopatológico es importante reconocerlas. Algunas de ellas se asocian con características clínicas o demográficas particulares o con una evolución más o menos agresiva. Sin embargo, esta división morfológica obedece más a la necesidad de aprender muchas cosas que ignoramos aún, pretende que denominemos las lesiones con una nomenclatura homogénea y que en un futuro próximo podamos acercarnos con más precisión a la etiología de la enfermedad.
La GEFyS puede presentarse a cualquier edad. Diferentes trabajos indican una predilección por el sexo masculino. Se encuentra en aproximadamente un 7 a 15% de casos de síndrome nefrótico en niños y 10 a 20% en adultos. En la mayoría de estudios hay una incidencia mayor de GEFyS en afroamericanos y descendientes de africanos. En este grupo de pacientes la GEFyS es la principal causa de síndrome nefrótico (36-80% de casos).
El pronóstico de la GEFyS es diferente del de la enfermedad de cambios mínimos, hay mayor incidencia de insuficiencia renal, muy poca respuesta a esteroides, y mayor incidenica de hematuria e hipertensión.
La etiología de la GEFyS está aún lejos de ser dilucidada, en gran parte debido a que esta enfermedad parece ser una expresión patológica de diferentes tipos de lesión. Por lo tanto, parecen haber varias (quizá muchas) causas de GEFyS. Entre los mecanismos patogénicos se ha postulado persistentemente la presencia de factores humorales circulantes; esta hipótesis está respaldada por la recurrencia de la enfermedad en riñones trasplantados y por la transferencia de un "factor de permeabilidad" en el suero de pacientes a animales de experimentación (Wilkinson AH et al, Clin Sci 77:43-8, 1989 [PubMed link]; Zimmerman SW, Clin Nephrol 22:32-8, 1984 [PubMed link]). Además, la respuesta a esteroides en algunos pacientes ha sugerido la participación del sistema inmune en la patogénesis. Sin embargo, nunca ha podido identificarse este supuesto factor. Algunos autores han informado que el receptor circulante de urokinasa ("serum soluble urokinase receptor -suPAR") puede ser un factor circulante que causa GEFyS (Wei C, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med. 2011:31;17(8):952-60. [PubMed link])
Factores hemodinámicos han sido también implicados. La hiperfiltración e hipertrofia glomerular han sido, experimentalmente, asociadas a lesión glomerular segmentaria; esto ayudaría a explicar la GEFyS en diabéticos y personas con masa renal disminuida. Este mecanismo se relaciona más con las formas secundarias de la GEFyS.
La lesión de células epiteliales glomerulares es otro mecanismo importante en algunas formas de GEFyS y se ha asociado principalmente con el uso de drogas intravenosas y con la infección por VIH.
Hay evidencia de factores genéticos en la patogénesis de la GEFyS y varias familias con la enfermedad han sido descritas, en ellas han sido detectadas diversas mutaciones (Sánchez de la Nieta MD, Arias LF, et al., Glomeruloesclerosis focal y segmentaria familiar. Nefrologia. 2003;23(2):172-6. [PubMed link / [Free full text]).
Por último, y quizá más importante, ha sido el descubrimiento, y hasta ahora entendimiento parcial, del complejo de proteínas que interactúan en el diafragma de hendidura del podocito ("slit diaphragm"), que aquí llamaremos, por ser un término más descriptivo de su estructura y función, diafragma de filtración. Los podocitos tienen un fenotipo arquitectural muy desarrollado y en particualar el diafragma de filtración es un tipo único de unión celular, en la cual las características de permeabilidad están determinadas por proteínas específicas. Como ya lo expresamos en el capítulo de enfermedad de cambios mínimos, un gran avance en el entendimiento del síndrome nefrótico idiopático se produjo con el descubrimiento, en 1998, de la nefrina (NPHS1), el gen mutado en el síndrome nefrótico congénito de tipo finlandés. Esto permitió establecer que el podocito es el componente central de la barrera de filtración. Poco después se descubrió el gene mutado en el síndrome nefrótico corticorresistente de inicio temprano: NPHS2 que codifica otra proteína exclusiva del podocito: la podocina. La podocina está estructuralmente relacionada con la familia de proteínas: estomatinas, que son proteínas transmembrana involucradas en el andamiaje del citoesqueleto. Se propone que la nefrina y la podocina interactúan a través de una unión a la proteína asociada a CD2 (CD2AP). Esta proteína hasta ahora había sido conocida como una molécula que liga el citoesqueleto de actina de las células T a zonas con las que contacta esta célula. La podocina y nefrina se unen al citoesqueleto del podocito (proteínas transmembrana) y su disrupción no sólo afecta el diafragma de filtración sino la integridad de todo el proceso podocitario. La podocina y la nefrina son expresadas tanto en la superficie celular como intracelularmente (D'Agati VD. Podocyte injury in focal segmental glomerulosclerosis: Lessons from animal models (a play in five acts). Kidney Int. 2008;73(4):399-406. [PubMed link]).
En el síndrome nefrótico se demuestran, cada vez con más frecuencia, alteraciones en esta compleja estructura de proteínas. Algunos autores han encontrado que la nefrina y podocina se localizan separadas del citoesqueleto en casos de síndrome nefrótico (Doublier S, et al., Am J Pathol 158:1723-31 [PubMed link / Free full text], 2001; Luimula P et al., Kidney Int 58:1461-8, 2000 [PubMed link]).
Se sabe que la nefrina, podocina y CD2AP están asociados, junto con la actina, en un complejo "lipid rafts" (traducido, de una manera aproximada, como: "lípidos balsa", o de transporte). Una de las funciones clave postuladas para los "lipid rafts" es facilitar transmisión rápida de señales, y la nefrina y podocina han mostrado ser interdependientes para iniciar una cascada de señales intracelulares, aunque las consecuencias de esto son aún desconocidas (Huber TB et al, Hum Mol Gen 12:3397, 2003 [PubMed link]; Coward R.J.M. et al, J Am Soc Nephrol 16:629, 2005 [PubMed link]).
Una revisión de las proteínas podocitarias y otras posibles etiologías de síndrome nefrótico en: Chen YM, Liapis H. Focal segmental glomerulosclerosis: molecular genetics and targeted therapies. BMC Nephrol. 2015 Jul 9;16:101. [PubMed link].
La identificación de un factor circulante como la molécula patogénica del síndrome nefrótico en enfermedad de cambios mínimos y en GEFyS ha sido, como lo expresan elegantemente Coward et al (J Am Soc Nephrol 16:629, 2005 [PubMed link]), un "santo grial" para los investigadores en este campo. Sin embargo, estos mismos autores proponen, en un muy bonito e interesantemente expuesto trabajo, que el plasma humano podría contener factores que son cruciales para la maduración y distribución normal de estas proteínas del complejo: hendiduras del diafragma-nefrina-podocina-CD2AP-citoesqueleto del podocito. Así, ha sido tentativamente hipotetizado que el síndrome nefrótico resultaría de la carencia de un factor que está normalmente presente en el plasma humano y no de la presencia de un factor circulante patogénico (Coward R.J.M. et al, J Am Soc Nephrol 16:629, 2005 [PubMed link]). Según estos autores, el síndrome nefrótico es un grupo heterogéneo de enfermedades y puede ser que en algunas, como la GEFyS, haya inicialmente una pérdida o imbalance de importantes factores plasmáticos que llevan a la disrupción de la integridad del diafragma de filtración y posterior pérdida de los procesos podocitarios. En otras nefropatías, como la nefritis lúpica, la disrupción generalizada del diafragma sería consecuencia de pérdida importante de proteínas.
Todo lo anterior ha llevado a la formulación de una hipótesis actualizada de la patogénesis del síndrome nefrótico. El podocito en su forma madura, y los procesos podocitarios con su diafragma de filtración, son constantemente mantenidos por un entorno de factores, algunos de los cuales son circulantes y algunos producidos localmente (autocrinos/paracrinos). La pérdida o alteración de este balance, sea primaria o secundaria, resulta en la disrupción de las señales transmitidas por la nefrina, llevando a pérdida de la estabilidad del diafragma y reorganización de los filamentos de actina, causando relocalización intracelular del complejo del diafragma y borramiento de los procesos podocitarios (Coward R.J.M. et al, J Am Soc Nephrol 16:629, 2005 [PubMed link]).
Aun así, queda mucho por saber y con toda seguridad conoceremos nuevos hallazgos y teorías sobre la etiología y patogénesis en los próximos años. En resumen: Se considera que la GEFyS primaria o idiopática está relacionada con lesión podocitaria. Se han propuesto varios factores circulantes que afectarían la barrera de permeabilidad de los podocitos, pero en ningun de ellos ha logrado demostrarse con certeza que causen GEFyS. Ésta también puede ser causada por alteraciones genéticas. Estos genes son principalmente los que regulan la estructura del diafragma de filtración, el citoesqueleto de actina de los podocitos o la estructura de los procesos podocitarios. El modo de herencia y la edad de inicio son diferentes según el gen implicado. Se ha destacado el papel de las células epiteliales parietales (PECs). Los podocitos y PEC tienen progenitores mesenquimales comunes, por lo tanto, las PEC podrían ser una fuente de repoblación de podocitos después de la lesión podocitaria. Las PEC activadas migran a las lesiones segmentarias y también pueden contribuir a la progresión de la esclerosis. Se pueden usar marcadores de activación de PEC, incluyendo CD44, para distinguir GEFyS de la enfermedad de cambios mínimos (Lim BJ, Yang JW, Do WS, Fogo AB. Pathogenesis of Focal Segmental Glomerulosclerosis. J Pathol Transl Med. 2016;50(6):405-410. [PubMed link]). .
En la clasificación etiológica de la GEFyS se debe diferenciar la primaria de las formas secundarias. La forma primaria (idiopática) es aquella en la que desconocemos, aunque existan diversas hipótesis, la etiología. Las formas secundarias se han dividido en (D`Agati V et al, Am J Kidney Dis 43:368, 2004 [PubMed link]):
1. Familiares/genéticas: Mutaciones en los genes de podocina, nefrina, alfa-actinina 4, beta integrina, etcétera.
2. Asociada a virus: VIH, parvovirus B 19.
3. Inducida por drogas: Heroina, interferón alfa, litio, pamidronato, etcétera.
4. Mediada por respuestas adaptativas estructurales-funcionales: A: masa renal reducida (agenesia, displasia renal, ablación quirúrgica, nefropatía por reflujo, nefropatía crónica del injerto, etcétera); y B: con masa renal normal (hipertensión, obesidad, procesos vaso-oclusivos, anemia de células falciformes, etcétera).
Ver Caso 87 de nuestra serie de casos: GEFS en paciente con anemia de células falciformes.
Clinica: La principal manifestación es proteinuria severa, usualmente con síndrome nefrótico completo. Aunque la proteinuria suele ser severa, hay un porcentaje de casos (alrededor del 20%) en los que la proteinuria es menor de 2,5 g/24h, muchos de estos pacientes presentarán síndrome nefrótico más tarde en la evolución de la enfermedad. Cuando hay proteinuria subnefrótica debe sospechase GEFyS secundaria, en estos casos la proteinuria suele ser progresiva. Algunos casos se presentan como proteinuria asintomática. La proteinuria tiende a ser no selectiva y en muchos casos es masiva: >10g/24h. La hematuria es una característica común, la mayoría de veces microscópica, pero ocasionalmente macroscópica. Con frecuencia hay hipertensión arterial y en algunos casos se detecta falla renal al momento del diagnóstico. No hay alteración de los niveles de complemento sérico.
Entre el 25% y el 60% de pacientes, de acuerdo a diferentes series, desarrollan insuficiencia renal terminal a los 10 años del disgnóstico. Pocos casos presentan respuesta a esteroides. En algunos casos se describe remisión sostenida y en otros hay persistencia de la proteinuria, pero sin alteración de la función renal.
Datos de laboratorio: Proteinuria en rango nefrótico en la mayoría de casos, hematuria micro en muchos de ellos. En una cuarta o quinta parte de los casos hay aumento de creatinina sérica y BUN. C3, C4 normales.
Histopatología
La característica histológica es esclerosis de segmentos del penacho en algunos glomérulos, con expansión del mesangio y colapso de luces capilares en estos segmentos. La lesión puede ser más pronunciada en el polo vascular o en la periferia del penacho. Al avanzar el proceso la esclerosis se torna global y es indistinguible de la esclerosis secundaria a otras enfermedades. Los segmentos de esclerosis son positivos con las tinciones de PAS y plata-metenamina (colágeno tipo IV). Desde los años cincuenta se ha dicho que los glomérulos yuxtamedulares son los más comprometidos por la lesión segmentaria. Los podocitos están, en muchas ocasiones, hipertróficos e hiperplásicos en la superficie del penacho que rodea la lesión esclerosante ("cap lesion" - lesión "en gorro") y con frecuencia aparecen con gotas de reabsorción proteica y lipídica. Los glomérulos sin lesiones esclerosantes pueden aparecer normales o con incremento de la celularidad mesangial y, a veces, hipertróficos (glomerulomegalia).
Figura 1. Penacho glomerular con esclerosis en la mitad superior; los segmentos de la mitad inferior presentan mesangio y paredes y luces capilares con arquitectura conservada. (Tricrómico de Masson, X400).

Figura 2. Con la plata-metenamina resaltan los segmentos de pérdida de la estructura capilar y esclerosis. Los podocitos que recubren estos segmentos presentan hipertrofia e hiperplasia. (Plata-metenamina, X400).
En algunos casos la lesión no tiene el aspecto de esclerosis (cicatrización por colágeno glomerular) sino de acúmulos homogéneos: Hialinos, que son acúmulos de material eosinofílico, cristalino, PAS positivo, fuschinofílicos o verdes con el tricrómico e idénticos a la lesión vista en diabetes mellitus. Este material es plata negativo, en contraste a los segmentos de esclerosis, y frecuentemente se acompaña de vacuolas lipídicas. Este material probablemente representa acúmulos de proteínas plasmáticas. Las lesiones hialinas usualmente acompañan a las lesiones esclerosas, pero hay casos en los que todas las lesiones son hialinas. Aunque parecen ser diferentes aspectos morfológicos de una misma enfermedad, podrían tratarse de fenómenos fisiopatogénicamente diferentes. Algunos autores, y así lo hacemos nosotros, diagnosticamos las lesiones sin hialinosis como: "GEFyS" y las que tienen hialinosis: "GEFyS con hialinosis", en un intento por diferenciar morfológicamente estos dos patrones y tratar de determinar si hay diferencias clínicas y/o etiopatogénicas.

Figura 3. Los segmentos hialinos son eosinofílicos, con aspecto homogéneo (flechas); se ven diferentes a los segmentos de esclerosis, pero, podrían corresponder a lesions en diferente estadio de evolución. (H&E, X400).

Figura 4. Hialinosis segmentaria con vacuolas lipídicas y color rojizo con el tricrómico (flecha). El penacho adyacente presenta solidificación, con disminución o pérdida de luces capilares. (Tricrómico de Masson, X400).
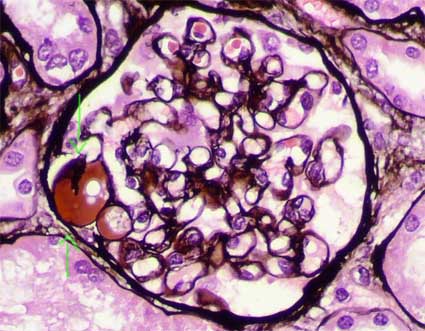
Figura 5. Los segmentos hialinos son negativos con la plata-metenamina y resaltan en medio del penacho. Los otros segmentos muestran paredes capilares y mesangio conservados. (Plata-metenamina, X400).
En los segmentos de esclerosis o de hialinosis, o rodeándolos, pueden haber células espumosas.

Figura 6. Células grandes, con citoplasma claro, vacuolado y núcleos pequeños, redondeados. Estas células son frecuentes en la GEFyS, pero pueden verse en cualquier síndrome nefrótico: Células espumosas. (Tricrómico de Masson, X1000).
De acuerdo a la localización de la lesión en el penacho, al grado de celularidad y al aspecto de las paredes capilares, se han clasificado cinco variantes morfológicas de GEFyS (ver más adelante).
Algunos autores consideran los casos con hipercelularidad mesangial difusa como una variante de GEFyS y la asocian con un curso más agresivo, sin embargo, otros trabajos no encuntran diferencia significativa respecto a la evolución clínica.
Es frecuente encontrar focos de atrofia tubular, fibrosis intersticial e infiltrado inflamatorio. La severidad del daño crónico túbulo-intersticial correlaciona bien, al igual que en otras glomerulopatías, con la severidad del daño renal y con la progresión a insuficiencia renal terminal. En los túbulos es frecuente encontrar, como en cualquier otra causa de síndrome nefrótico, gotas de reabsorción PAS positivas. Con frecuencia hay algún grado de fibrosis intimal arterial y de arterioloesclerosis hialina, principalmente en adultos.
Inmunofluorescencia
Los glomérulos sin lesión y los segmentos del penacho que no presentan esclerosis o hialinosis no muestran depósito de inmunoglobulinas o fracciones del complemento. En los segmentos de lesión se identifica, con mucha frecuencia, depósitos de IgM y de C3. Se cree que estos depósitos no representan complejos inmunes ni son patogénicos y que son el resultado de atrapamiento de proteínas plasmáticas. Ocasionalmente se identifican depósitos poco intensos de IgG en los segmentos de lesión. En el citoplasma de podocitos y de células tubulares pueden identificarse Igs. y complemento que representan reabsorción de proteínas filtradas al espacio urinario.

Figura 7. La inmunofluorescencia muestra depósitos igualmente focales y segmentarios de IgM y de C3; estos depósitos probablemente no son patogénicos, sino, atrapamiento en lesiones esclerosantes o hialinas. (Inmunofluorescencia directa con anticuerpos anti-IgM, marcados con fluoresceina, X400).
Microscopía electrónica
Los segmentos de esclerosis muestran incremento de la matriz mesangial y de material similar al de la membrana basal. Los segmentos hialinos se ven homogéneos, electrón densos y, contrario a depósitos inmunes, tienen un borde mal definido, sin la nitidez que muestran los inmunocomplejos; además, el material hialino se encuentra en áreas de esclerosis o de colapso capilar. Los glomérulos sin lesiones segmentarias pueden presentar borramiento de procesos podocitarios en una extensión variable. La célula epitelial puede encontrarse desprendida de la membrana basal glomerular (MBG) dejando áreas claras; la severidad de este cambio se ha asociado con la naturaleza no selectiva de la proteinuria (Jennette JC, et al. (Eds.). Heptinstall`s Pathology of the Kidney, 5º ed., Lippincott-Raven, Philadelphia, 1998, p.218). Es común la transformación microvellosa del citoplasma podocitario y una apariencia laminada de la MBG. Se observan vacuolas lipídicas intracitoplasmáticas.
Variantes histológicas de la GEFyS
El espectro histopatológico de la GEFyS es amplio y complejo y requiere una clasificación basada en consenso de expertos investigadores en el tema con el fin de lograr una nomenclatura homogénea. Aunque hay considerable controversia respecto al significado y definición de estas variantes, la clasificación intenta definir más precisamente conceptos que permitan un mismo lenguaje entre todos los nefropatólogos del mundo. Esta clasificación morfológica incluye casos de enfermedad primaria y secundaria, pero excluye cualquier lesión glomerular que sea consecuencia de otra glomerulopatía. Aún queda por saber si estos patrones morfológicos conllevan implicaciones significativas con respecto a la etiología, presentación o evolución clínica, patogénesis, pronóstico o tratamiento óptimo (D'Agati VD, et al, Am J Kidney Dis. 2004;43:368-82 [PubMed link]). Más importante que esta clasificación histopatológica (no muy valorada por algunos) es intentar buscar la etiología o factores asociados en cada caso.
GEFyS sin otra especificación (NOS)
Es la forma más común. En esta variante hay lesiones esclerosantes segmentarias que pueden comprometer cualquier parte del penacho, pero, por definición, deben descartarse las otras cuatro variantes, es decir, esta variante se diagnostica por descarte. En esta variante pueden haber lesiones hialinas, hipercelularidad mesangial, hipertrofia-hiperplasia de podocitos o glomerulomegalia. Todas las otras variantes pueden evolucionar a esta forma de GEFyS.
GEFyS Variante perihiliar
Para diagnosticar esta variante debemos descartar la variante hipercelular y la variante colapsante. En otras palabras, si hay muchos glomérulos con lesiones perihiliares, pero al menos uno con lesion hipercelular o con lesión colapsante, no debe diagnosticarse como perihiliar, sino como estas últimas respectivamente. Si hay algún glomérulo con "Tip lesion" no excluye esta categoría.
Para hacer el diagnóstico de esta variante se requieren dos requisitos: 1.) Al menos un glomérulo con hialinosis perihiliar, acompañada o no de esclerosis; y 2.) Más del 50% de glomérulos con lesiones segmentarias deben tener esclerosis y/o hialinosis perihiliar.
Otros glomérulos pueden presentar lesiones como en GEFyS (NOS). Puede haber algun grado de proliferación mesangial y pueden identificarse vacuolas lipídicas e hipertrofia o hiperplasia de podocitos, aunque este último hallazgo es menos frecuente que en otras variantes.
Este tipo de lesión es la que más se ha relacionado con formas secundarias asociadas a respuestas adpatativas del glomérulo, a pérdida de masa renal o a hipertensión glomerular (obesidad, falla cardíaca congénita, oligomeganefronia, nefropatía por reflujo, etcétera). Tambien puede encontrarse esta variante en GEFyS primaria.

Figura 8. En esta microfotografía, a la izquierda (flechas azules) vemos una arteriola glomerular, el penacho, a la derecha, muestra segmentos hialinos en su polo vascular (flechas verdes). La variante perihiliar se carcateriza por presentar al menos un glomérulo con hialinosis perihiliar (como en esta imágen), acompañada o no de esclerosis, y más del 50% de glomérulos con lesiones segmentarias deben tener esclerosis y/o hialinosis perihiliar. No deben haber glomérulos con lesiones colapsantes ni hipercelulares. (Tricrómico de Masson, X400).
Ver el Caso 168 de nuestra Serie de Casos: Variante perihilar de GEFS
GEFyS variante celular
Para poder diagnosticar esta variante debe descartarse la variante "Tip" y la variante colapsante; en otras palabras, si hay al menos un glomérulo con "tip lesión" o con características de colapsante, se excluye esta variante.
Se define por la presencia de al menos un glomérulo con hipercelularidad endocapilar que compromete al menos el 25% del penacho y ocluye las luces capilares. Puede afectarse cualquier segmento perihiliar o periférico. Las células endocapilares incluyen células endoteliales, macrófagos y células espumosas. También pueden identificarse polimorfos y linfocitos. Puede haber cariorrexis, picnosis, depósitos hialinos y apoptosis. Algunas veces puede haber fibrina, pero no debe identificarse ruptura de la MBG (en estos casos sospechar un glomerulonefritis necrotizante). Puede haber hipertrofia/hiperplasia de podocitos y sinequias a la cápsula de Bowman. En otros glomérulos pueden haber lesiones de GEFyS (NOS).
Cuando se observa al menos una lesión "Tip" debe descartarse la variante celular; esta exclusión es basada en la observación de que muchas "Tip lesion" son celulares.
En muchos textos se denomina lesion celular a la hiperplasia de podocitos. Esta alteración no debe confundirse con la variante celular en la clasificación actual.
En mi opinión, esta variante es algo confusa, dificil de diagnosticar y algunos autores no registran casos en sus series. Desde el punto de vista pronóstico, no parece haber diferencias con la variante NOS. Yo sólo la diagnostico en casos de síndrome nefrótico completo, inmunofluorescencia indudablemente negativa (o IgM y/o C3 en lesiones segmentarias - inespecífica) y luego de un exhaustivo análisis para descartar otras glomerulopatías.

Figura 9. Las flechas señalan un segmento con incremento de la celularidad y disminución o pérdida de las luces capilares; la hipercelularidad se debe a proliferación de células intrínsecas del glomérulo y células inflamatorias que han migrado al penacho, en este caso mononucleares (linfocitos y monocitos). En ocasiones podemos encontrar polimorfos. En el caso de la microfotografia encontramos lesiones esclerosantes segmentarias y focales de tipo NOS en 4 de 18 glomérulos y sólo uno (el de la foto) con carcaterísticas de variante hipercelular. (H&E, X400).
GEFyS variante "Tip"
El extremo del penacho glomerular adyacente al origen del túbulo proximal es llamado extremo "tip", o, traducido al castellano, extremo "de la punta". Así que esta variable podríamos traducirla al castellano como: "Variante de lesión de la punta". Sin embargo, por comodidad y por una particular y subjetiva apreciación estética, preferimos llamarla "variante tip".
Para diagnosticar esta lesión debemos excluir la variante colapsante, es decir, si hay al menos un glomérulo con las características de esta última, se descarta la variante "tip". Además, si hay lesiones en localización perihiliar se descarta la variante tip, teniendo en consideración la naturaleza periférica de las lesiones en la "tip".
Se define por la presencia de al menos una lesión comprometiendo el dominio tip: El 25% del penacho adyacente al origen del túbulo proximal. En esta lesión pueden evidenciarse adherencias del penacho a la cápsula de Bowman del cuello del túbulo o cerca de éste, o adherencias en la luz del túbulo; confluencia de podocitos en el sitio de lesión; segmentos hialinos; segmentos con hipercelularidad endocapilar (<50% del penacho); segmentos de esclerosis (<25% del penacho). Comunmente se encuentran células espumosas y podocitos con hipertrofia e hiperplasia. Puede haber hipercelularidad mesangial. En otros glomérulos pueden haber lesiones esclerosantes o hipercelularidad endocapilar en sitios diferentes al dominio tip, sin embargo, no deben comprometer segmentos perihiliares.

Figura 10. En este glomérulo podemos apreciar la localización carcaterística de las lesiones "tip" (de la punta glomerular). Podemos ver sinequias y esclerosis (como en este caso), o podemos ver segmentos hialinos o hipercelularidad endocapilar comprometiendo esta porción del penacho. (Tricrómico de Masson, X400).

Figura 11. En este glomérulo la lesión "tip" se caracteriza por sinequia del penacho a la cápsula de Bowman cercana al cuello del túbulo proximal. (Plata-metenamina, X400).
Esta variante ha sido asociada, en diferentes trabajos, con un mejor pronóstico (menor riesgo de falla renal terminal) (Howie AJ, et al. Glomerular tip changes in childhood minimal change nephropathy. Pediatr Nephrol. 2008 Aug;23(8):1281-6. [PubMed link]). En nuestra serie de pacientes hispánicos, nosotros no encontramos un pronóstico "favorable" en esta variable y la evolución clínica fue casi similar a GEFyS variante NOS (Arias LF, et al. Tip variant of focal segmental glomerulosclerosis: outcome and comparison to 'not otherwise specified' variant. Nephrol Dial Transplant. 2011;26(7):2215-21. [PubMed link]). Es mi opinión que esta variante no debería considerarase "benigna".
Ver Caso 9 y Caso 49 ("tip lesion") de nuestra serie de casos
GEFyS variante colapsante (muchos consideramos esta glomerulopatía como una enfermedad glomerular diferente a GEFyS, no una variante de ésta)
Esta categoría excluye todas las otras variantes. Se define por el compromiso de al menos un glomérulo con colapso capilar y notoria hiprertrofia e hiperplasia de podocitos. Las paredes capilares presentan retracción y colapso. Las lesiones pueden ser segmentarias o globales y comprometer segmentos periféricos o perihiliares. El número de glomérulos afectados es muy variable. Es inusual encontrar sinequias a la cápsula de Bowman y lesiones hialinas. En otros glomérulos pueden encontrarse lesiones de cualquier categoría: esclerosis, hipercelularidad endocapilar, tip lesión o lesiones esclerosantes globales.

Figura 12. Observe todo el penacho colapsado, sin luces capilares bien conservadas, con aspecto irregular y "arrugado" de las paredes capilares y con una marcada hipertrofia e hiperplasia de podocitos que acompaña este tipo de lesions colapsantes. Este caso corresponde al de un paciente de 37 años con síndrome nefrótico, HIV negativo y sin otros factores asociados: GEFyS colapsante primaria (idiopática). (Plata-metenamina, X400).

Figura 13. Las lesiones colapsantes puden no ser globales y comprometer sólo algunos segmentos del penacho. Aunque en este caso la lesión es global, observe mayor colapso y mayor hipertrofia e hiperplasia de podocitos en los segmentos señalados con flechas (Plata-metenamina, X400).
Este patrón ha sido asociado con GEFyS primaria, asociada a VIH, toxicidad por pamidronato, parvovirus B 19, factores hemodinámicos y otras raras asociaciones (Albaqumi M, Barisoni L. Current Views on Collapsing Glomerulopathy. J Am Soc Nephrol. 2008;19(7):1276-81. [PubMed link]; Gulati A, et al. Idiopathic collapsing glomerulopathy in children. Clin Exp Nephrol. 2008;12(5):348-53. [PubMed link])
En pacientes con infección por VIH y esta variante, es frecuente encontrar inclusiones túbuloreticulares en células endoteliales (microscopía electrónica) que no se identifican en otras lesiones colpsantes no asociadas a VIH, y que también puede identificarse en otras enfermedades glomerulares como la nefritia lúpica.
Esta variante parece tener un curso más agresivo, con proteinuria más severa y más rápida evolución a falla renal terminal. Se ha encontrado mayor predilección por pacientes afroamericanos.
La glomerulopatía Colapsante (GC) puede ser una enfermedad primaria (no asociada a otras alteraciones o factores), o puede estar asociada a infección por el VIH, parvovirus B19, pamidronato, enfermedades autoinmunes, otros medicamentos, otras infecciones, neoplasias, trastornos genéticos, ateroembolia , trastornos vaso-oclusivos agudos y otras asociaciones aún menos frecuentes. La GC no es sólo una alteración glomerular o enfermedad de podocitos, todas las células epiteliales renales puede verse afectadas. El grado de daño tubulointersticial varía de un caso a otro y parece ser más pronunciado en las formas en que el daño celular epitelial intrínseco es el factor relacionado, como una infección viral (Albaqumi M, Barisoni L. Current views on
collapsing glomerulopathy. J Am Soc Nephrol. 2008;19(7):1276-81.
[PubMed
link]). Los intentos de identificar mecanismos patogénicos en GC han planteado una serie de hipótesis, sin embargo, los mecanismos por los que estos trastornos conducen a lesiones podocitarias son poco conocidos. Algunos investigadores han centrado su atención en la mitocondria. Las mutaciones en los genes que codifican proteínas implicadas en la cadena respiratoria mitocondrial pueden conducir a GC en ratones y seres humanos. Dado el papel crucial de la disponibilidad de oxígeno en el metabolismo de las mitocondrias, estos datos sugieren que los podocitos pueden ser muy sensibles a las condiciones de hipoxia. Es muy llamativa la evidencia reciente de que la sobre-regulación del factor inducible por hipoxia en podocitos lleva su proliferación y desdiferenciación en la nefropatía asociada al VIH. Podocitos lesionados muestran la pérdida de algunos marcadores de podocitos, la inducción de la expresión de algunas moléculas y transdiferenciación a linaje de macrófagos (Canaud G, et al. Glomerular collapse associated with subtotal renal infarction in kidney transplant recipients with multiple renal arteries.Am J Kidney Dis. 2010 Mar;55(3):558-65. [PubMed link]).
El término "glomerulopatía colapsante" fue utilizado por primera vez en 1986, y se supone que es una variante de GSF. Sin embargo, la GC es frecuentemente una enfermedad agresiva, con proteinuria masiva y enfermedad renal rápidamente progresiva. La evolución notablemente más agresiva en GC que en las otras variantes de GEFS y las características fenotípicas de los podocitos que sugiere una patogénesis diferente, han llevado a algunos autores a proponer que la GC no es una variante del GEFS (Howie AJ. Problems with 'focal segmental glomerulosclerosis'. Pediatr Nephrol. 2011;26(8):1197-205. [PubMed link]).
Ver Caso 6 - Caso 40 - Caso 86 - Caso 159 (glomerulopatías colapsantes) de nuestra serie de casos.
Bibliografía