
Homepage - - - Tutorial Index
Focal and segmental Glomeruloesclerosis
Focal and segmental glomerulosclerosis (FSGS) is a disease characterized
morphologically by segments of sclerosis in some glomeruli. It can be primary
or secondary and it usually present as nephrotic syndrome (NS). The glomeruli
without segmental lesions can display alterations of the mesangial cellularity
and/or loss (“effacement”) of podocyte foot process.
The term “sclerosis” means healing and it is characterized by accumulation of glomerular collagen (type IV). Nevertheless, in FSGS some of the segmental lesions are not sclerosis, but hyaline deposits: Hyalinosis. This feature originated the classical denomination, more of the French school, focal and segmental hyalinosis. Other investigators designate those cases in which there are hyaline segments: “FSGS with hyalinosis”. There is no a clear explanation why in some cases there are hyaline segments and in other no. Some authors consider that hyaline lesions are precursory of those sclerosing; nevertheless, seems that in many cases sclerosing lesions begins thus, without a phase of hyalinosis.
FSGS is a clinicopathologic entity. In some renal biopsies we can demonstrate segmental and focal sclerosing lesions that can be secondary to previous episodes of focal glomerulonephritis (GN); these cases are different to true FSGS, although in many cases differential diagnosis is not an easy task.
The histologic features of FSGS do not allow differentiating the primary forms from the secondary ones. In order to make this differentiation we must help us with clinical findings and laboratory data, and it is very important evaluate alterations in other histologic compartments: vessels, interstitium, and tubules. Even so, we will not be able to do, in many cases, a clear distinction between primary and secondary forms. In the future, when we will have more knowledge of the etiology and physiopathogenesis of the disease, we will be able to distinguish better the different forms from the disease. It is possible that in the future we can diagnose many of these cases according to etiology or pathogeny, and perhaps the FSGS term will become obsolete (Cameron JS, The enigma of focal segmental glomerulosclerosis. Kidney Int, Suppl 57, S119, 1996 [PubMed link]).
There are morphologic variants of FSGS and for the histopathologic diagnosis it is important to recognize them. Some of them are associated with clinical characteristics or demographic aspects, or a more or less aggressive evolution. Nevertheless, this morphologic division obeys more to the necessity to learn many things that we still ignore, tries that we denominate the lesions with a homogenous nomenclature, and will be very important to determinate particular etiologic or pathogenic mechanisms.
FSGS can appear to any age. Diverse works indicate a predilection by masculine sex. FSGS is the cause of NS in approximately 7% to 15% in children and 10% to 20% in adults. In most of studies there is a greater incidence in Afro-American and descending of African; in this group of patients FSGS is the main cause of NS (36-80% of cases). In our series of Hispanic patients FSGS is the more frequent glomerulopathy: 31% of all primary glomerular diseases (unpublished data).
The prognosis of FSGS is different to minimal change disease (MCD): there is greater incidence of chronic renal failure, very little response to steroids, and greater incidence of hematuria and hypertension.
The etiology of FSGS is still far from being explained, to a great extent because this disease seems to be a pathological expression of different types of injury. Therefore, it seem to have several (perhaps many) causes of FSGS. One of the postulated pathogenic mechanisms implicates the presence of circulating humoral factors; this hypothesis is supported by the recurrence of the disease in transplanted kidneys and by the transference of a “factor of permeability” in the serum of patients to experimentation animals (Wilkinson AH et al, Clin Sci 77:43-8, 1989 [PubMed link]; Zimmerman SW, Clin Nephrol 22:32-8, 1984 [PubMed link]). In addition, response to steroids in some patients has suggested the participation of the immune system in the pathogenesis. Nevertheless, this factor has been never found. Some authors have reported that circulanting (serum soluble urokinase receptor -suPAR) may be a circulating factor causing FSGS (Wei C, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med. 2011:31;17(8):952-60. [PubMed link])
Hemodynamic factors have also been implicated. Glomerular hypertrophy and hyperfiltration have been, experimentally, associated to segmental glomerular lesions; this would help to explain the frequency of FSGS in diabetics and people with diminished renal mass. This mechanism is more related to secondary forms of FSGS.
Injury of tuft epithelial cells (podocytes) is another mechanism implicated in some forms of FSGS and it has been associated mainly to disease in intravenous drug abusers and HIV infection.
There is evidence of genetic factors in the pathogenesis of FSGS and several families with the disease have been described, in them have been located diverse mutations (Sánchez de la Nieta MD, Arias LF, et al., Glomeruloesclerosis focal y segmentaria familiar. Nefrologia. 2003;23(2):172-6. [PubMed link / [Free full text]).
Finally, and perhaps more important, it has been the discovery, and until now partial understanding, of the protein complex that interacts in the slit diaphragm of podocytes. Podocytes have an architectural phenotype very developed and in particular the slit diaphragm; it is a unique type of cellular union, in which the permeability characteristics are determined by specific proteins. The podocyte slit diaphragm has an important and direct role in glomerular filtration. Some of its protein components are involved in the mechanism of proteinuria. These proteins form a complex that contributes to its structure, connects the diaphragm to the intracellular actin cytoskeleton, and participates in signaling related to turnover of the glomerular filter. Most of these proteins are essential for a functional slit diaphragm and glomerular filtration, since mutation or inactivation of the corresponding genes causes proteinuria.
A great advance in the understanding of the idiopathic NS syndrome took place with the discovery, in 1998, of the nephrin (NPHS1), the muted gene in the congenital NS of Finnish type. This allowed establishing that the podocyte is the central component of the filtration barrier. Shortly after the muted gene in the autosomal recessive steroid resistant NS was discovered: NPHS2 that codifies another exclusive protein of the podocyte: podocin. The podocin is structurally related to the protein family: stomatins, that are transmembrane proteins implied in the scaffolding of the cytoskeleton. It is proposed that nephrin and podocin interact with union to the protein associated to CD2 (CD2AP). This protein until now had been well-known like a molecule that binds the cytoskeleton of actin of T-cells to zones with which this cell contacts. Podocin and nephrin are united to the cytoskeleton of the podocyte (transmembrane proteins) and their disruption not only affects the slit-diaphragm but the integrity of all the foot processes. Podocin and nephrin are expressed in both: cellular surface and intracellularly (D'Agati VD. Podocyte injury in focal segmental glomerulosclerosis: Lessons from animal models (a play in five acts). Kidney Int. 2008;73(4):399-406. [PubMed link])
In NS there are an increasing number of alterations in this complex protein structure. Some authors have found that nephrin and podocin are located separated of the cytoskeleton in cases of NS (Doublier S, et al., Am J Pathol 158:1723-31 [PubMed link / Free full text], 2001; Luimula P et al., Kidney Int 58:1461-8, 2000 [PubMed link]).
Nephrin, podocin, CD2AP and actin are associated in a complex with lipid rafts. One of the postulated key functions for lipid rafts is to facilitate fast transmission of signals, and nephrin and podocin have shown to be interdependent to initiate a cascade of intracellular signals, although the consequences of this are still not known (Huber TB et al, Hum Mol Gen 12:3397, 2003 [PubMed link]; Coward R.J.M. et al, J Am Soc Nephrol 16:629, 2005 [PubMed link]).
A revision of podocitary proteins and other factors related to nephrotic syndrome in: Chen YM, Liapis H. Focal segmental glomerulosclerosis: molecular genetics and targeted therapies. BMC Nephrol. 2015 Jul 9;16:101. [PubMed link]..
The identification of a circulating factor as the pathogenic molecule of the NS in minimal change disease and FSGS has been, as expressed by Coward et al (J Am Soc Nephrol 16:629, 2005 [PubMed link]), “a holy grail” for investigators in this field. Nevertheless, these same authors propose, in a very interesting work, that the human serum could contain factors that are crucial for development and normal distribution of these proteins of the complex: slit diaphragm-nephrin-podocin-CD2AP-podocyte cytoskeleton. Thus, it has been tentatively hypothesized that the NS would be originated by deficiency of a factor that is normally present in the human serum and not by the presence of a pathogenic circulating factor (Coward R.J.M. et al, J Am Soc Nephrol 16:629, 2005 [PubMed link]). According to these authors, NS is a heterogeneous group of diseases and can be that in some, like FSGS, there is initially a loss or imbalance of important serum factors that produce disruption of the integrity of the slit diaphragm and later loss of the podocyte foot processes. In other nephropathies, like lupus nephritis, generalized disruption of the slit diaphragm would be consequence of important loss of proteins.
All the previous information has allowed the formulation of an updated hypothesis of the pathogenesis of the NS. The mature podocyte and the foot processes with the slit diaphragm are constantly maintained by surroundings factors, some of which are circulating and some locally produced (autocrine/paracrine). The loss or alteration of this balance (can be by primary or secondary) can cause disruption of signals transmitted by the nephrin or other slit diaphragm-associated proteins, producing loss of the stability of the diaphragm and reorganization of actin filaments, causing intracellular relocalization of the protein complex and effacement of foot processes (Coward R.J.M. et al, J Am Soc Nephrol 16:629, 2005 [PubMed link]).
Even so, much remains to know and it is sure that we will know new findings and theories on the etiology and pathogenesis of FSGS in the next years, but in brief: Primary or idiopathic FSGS is considered to be related to podocyte injury. Several circulating factors affecting podocyte permeability barrier have been proposed, but not proven to cause FSGS. FSGS may also be caused by genetic alterations. These genes are mainly those regulating slit diaphragm structure, actin cytoskeleton of podocytes, and foot process structure. The mode of inheritance and age of onset are different according to the gene involved. The role of parietal epithelial cells (PECs) has been highlighted. Podocytes and PECs have common mesenchymal progenitors, therefore, PECs could be a source of podocyte repopulation after podocyte injury. Activated PECs migrate along adhesion to the glomerular tuft and may also contribute to the progression of sclerosis. Markers of activated PECs, including CD44, could be used to distinguish FSGS from minimal change disease. The pathogenesis of FSGS is very complex (Lim BJ, Yang JW, Do WS, Fogo AB. Pathogenesis of Focal Segmental Glomerulosclerosis. J Pathol Transl Med. 2016;50(6):405-410. [PubMed link]).
In the etiologic classification of FSGS is important differentiate primary forms from secondary forms. The primary form (idiopathic) is that in that we do not know, although exist diverse hypotheses, the etiology. The secondary forms have been divided in (D`Agati V et al, Am J Kidney Dis 43:368, 2004 [PubMed link]):
1.Familial / genetics: Mutations in genes of the podocin, nephrin, alpha-actinin-4, beta-integrin, TRPC6, an others.
2. Virus-associated: HIV, parvovirus B-19.
3. Induced by drugs: Heroin, interferon alpha, lithium, pamidronate, and others.
4. Mediated by adaptive structural-functional responses: A: reduced renal mass (agenesis, renal dysplasia, reflux nephropathy, surgical ablation, chronic allograft nephropathy, and so on); and B: with normal renal mass (vaso-occlusive processes, hypertension, obesity, sickle cell anemia).
See Case 87 of our case series: FSGS in a patient with sickle cell anemia.
Clinic features: The main manifestation is severe proteinuria, usually with complete NS. Although proteinuria usually is severe, there are a percentage of cases (around 20%) in which proteinuria is less than 2.5 g/24h, many of these patients will present later NS. When there is subnephrotic proteinuria, secondary FSGS should be suspected, in these cases proteinuria is usually progressive. Some cases appear like asymptomatic proteinuria. Proteinuria tends to be nonselective and in many cases it is massive: >10g/24h. There is microscopic hematuria, but occasionally it is macroscopic. Frequently there is arterial hypertension and in some cases renal failure at the time of diagnosis is detected. There is not serum complement levels alteration.
Between 25% and 60% of patients, according to different series, develop terminal renal insufficiency to 10 post-diagnosis years. In few cases there is response to steroids. In some cases maintained remission is described, and in others there is persistence of proteinuria, but without alteration of the renal function.
Laboratory features: Proteinuria in nephrotic range in most of cases, microhematuria in many of them. In 20-25% of cases there is serum creatinine and BUN increase. Normal C3 and C4 levels. In many cases (NS) dyslipidemia and hypoalbuminemia.
Histopathology
The characteristic histologic feature is sclerosis
of segments of the glomerular tuft in some glomeruli (“segmental and focal”),
with mesangial expansion and capillary lumen loss in these segments. The injury
can be more notorious in the vascular pole or in the periphery of the tuft.
When advancing the process the sclerosis becomes global and is indistinguishable
of the secondary sclerosis to other diseases. The sclerosing segments are positive
with PAS and silver-methenamine stains (type IV collagen). From the Fifties
it is said that juxtamedullary glomeruli are more compromised by the segmental
lesions. There are, in many cases, podocyte hypertrophy and hyperplasia, mainly
on the surface of the sclerosed tuft segment (“cap lesion” or “cellular
lesion”). Podocytes frequently appear with protein droplets and lipid
resorption. The glomeruli without sclerosing lesions can appear normal or with
increase of the mesangial cellularity and, sometimes, hypertrophic (glomerulomegaly).
.
Figure 1. Glomerular tuft segmental sclerosis in the superior half; segments in the inferior half display mesangium and capillary walls and lumens with conserved architecture. (Masson’s trichrome, X400).

Figure 2. With methenamine-silver stain the segments with loss of the capillary structure and sclerosis are better seen. The podocytes that cover these segments present hypertrophy and hyperplasia. (Methenamine-silver, X400).
In some cases the lesions do not have the aspect of sclerosis (healing by glomerular collagen) but appear as homogenous material: Hyaline deposits. They are constituted by crystalline, eosinophilic, PAS positive, and fuchsinophilic or blue with trichrome stains material; this material is similar to glomerular material found in diabetic nephropathy. This material is negative with silver stains, in contrast to sclerosed segments, and frequently it is accompanied by lipid vacuoles. This material probably represents accumulation of serum proteins. Hyaline lesions usually accompany sclerosing lesions, but there are cases in which all the lesions are hyaline. Although they seem to be different morphologic aspects from a same disease, could have a different physiopathogeny. Some authors, and thus we do, diagnose the cases without hyalinosis as: “FSGS” and those that have hyalinosis: “FSGS with hyalinosis”, in an attempt to differentiate morphologically these two patterns and to try to determine if there are clinical and/or etiopathogenic differences.

Figure 3. The hyaline segments are eosinophilic, with homogenous aspect (arrows); they are different to sclerosing segments, but they could correspond to lesions in different stage of evolution. (H&E, X400).

Figure 4. Segmental Hyalinosis with lipid vacuoles and reddish color with the trichrome stain (arrow). The adjacent tuft presents solidification, with diminution or loss of capillary lumens. (Mason’s trichrome, X400).
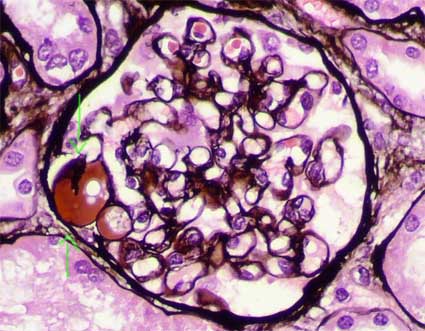
Figure 5. The hyaline segments are negative with the methenamine-silver stain. The other segments show conserved capillary walls and mesangium. (Methenamine-silver, X400).
Segments with sclerosis or hyalinosis, or in areas surrounding them, may have foam cells.

Figure 6. Large cells, with clear or vacuolated cytoplasm and small nuclei. These cells are frequent in FSGS, but they can be seen in any glomerulopathy causing NS: “foam cells”. (Masson’s trichrome, X1000).
According to the location of the lesion in the tuft, to the cellularity degree, and the aspect of the capillary tuft, five morphologic variants of FSGS have been proposed (see below).
Some authors consider the cases with diffuse mesangial hypercellularity as a variant of FSGS and associate it with a more aggressive course, nevertheless, other works do not find significant difference with respect to the clinical evolution.
It is frequent to find areas with tubular atrophy, inflammatory interstitial infiltrates and fibrosis. The severity of the tubule-interstitial chronic damage correlates well, like in other glomerulopathies, with the severity of the renal damage and with the progression to terminal renal failure. In tubules is frequent to find, like in any other cause of NS, PAS positive resorption droplets. Frequently there is some degree of arterial intimal fibrosis and hyaline arteriolosclerosis, mainly in adults.
Immunofluorescence
Glomeruli or segments without lesions do not
show immunoglobulins or complement deposition. In the segments with lesion is
identified, very frequently, deposits of IgM and C3. It is possible that these
deposits do not represent immune complexes nor they are pathogenic and that
they are the result of unspecific deposition of serum proteins. Occasionally
there are weak deposits of IgG in segments with lesions. In the cytoplasm of
podocytes and tubular cells we may dentify Igs. and complement that represents
resorption of filtered proteins.

Figure 7. The immunofluorescence shows equally focal and segmental deposits of IgM and C3; these deposits probably are not pathogenic, but they correspond to unspecific deposition of serum proteins. (Direct immunofluorescence with antibodies anti-IgM, marked with fluorescein, X400).
Electron microscopy
The sclerosed segments show increase of the mesangial matrix and material similar to the one of the basal membrane. The hyaline segments are homogenous, electron dense and, in opposition to immune deposits, they have a not well-defined edge, without the clearness that show the immunocomplexes; in addition, the hyaline material can be seen in areas with sclerosis or capillary collapse. Glomeruli without segmental lesions show “effacement” or “fusion” of podocyte foot processes in a variable extension. The epithelial cell may become detached from the glomerular basement membrane (MBG) leaving clear areas; the severity of this change has been associated with the nonselective nature of proteinuria (Jennette JC, et al. (Eds.). Heptinstall`s Pathology of the Kidney, 5º ed., Lippincott-Raven, Philadelphia, 1998, p.218). Microvillous transformation of the podocitary cytoplasm is common and a laminated appearance of the MBG may develop at the site of detachment. Intracytoplasmic lipid vacuoles are observed.
Histologic variants of FSGS
The histopathologic spectrum of FSGS is ample and complex and requires a classification based on consensus of experts dealing with the theme with the purpose of obtaining a homogenous nomenclature. Although there is considerable controversy with respect to the meaning and definition of these variants, the classification tries to define concepts than allow a same language among all the nephropathologists and nephrologists of the world. This morphologic classification includes cases of primary and secondary diseases, but it excludes any glomerular change that it is consequence of another glomerulopathy. Still it is to know if these morphologic patterns have significant implications with respect to the etiology, clinical features, outcome, pathogenesis, prognosis, or optimal treatment (D'Agati VD, et al, Am J Kidney Dis. 2004;43:368-82 [PubMed link]). More important than this histopathological classification (not very valued by some) is to try to find the etiology or associated factors in each case.
FSGS not otherwise specified (NOS)
The form is the commonest. In this variant there are segmental sclerosing lesions that can compromise any part of tuft, but, by definition, this category requires that all other categories (perihilar, cellular, tip, and collapsing) be excluded. It is defined by focal and segmental consolidation of the tuft by increased extracellular matrix, obliterating the glomerular capillary lumen. There may be segmental glomerular capillary wall collapse without overlying podocyte hyperplasia. Lesions of sclerosis are typically discrete and can affect perihiliar and/or peripheral segments. Lesions can be sclerosing or hyaline. There can be mesangial hypercellularity, podocyte hypertrophy-hyperplasia, or glomerulomegaly. All the other variants can evolve to this category of FSGS.
FSGS perihilar variant
In order to diagnose this variant we must exclude the hypercellular variant and the collapsing variant. In other words, if there are many glomeruli with perihilar lesions, but at least one with hypercellular or collapsing lesion, we do not diagnose the perihilar variant, but like these last ones respectively. If there are a glomerulus with “tip lesion” it does not exclude this category.
Defining criteria include both of the following: a) there must be at least 1 glomerulus with perihilar hyalinosis, with or without sclerosis; and b) more than 50% of glomeruli with segmental lesions must have perihilar sclerosis and/or hyalinosis.
Other glomeruli may show lesions as described in FSGS NOS. In some cases there is some degree of mesangial proliferation, and lipid vacuoles and hypertrophy or hyperplasia of podocytes may be identified, although this last finding is less frequent than in other variants.
This type of lesion is common in patients with secondary forms of FSGS mediated by an adaptive response to nephron loss or glomerular hypertension: in association with obesity, cyanotic congenital heart disease, reflux nephropathy, renal agenesis, or any renal disease with reduced number of functioning nephrons.

Figure 8. In this microphotography, we see a glomerular arteriole (blue arrows), in the tuft there are hyaline segments in its vascular pole (green arrows). The perihilar variant characterize by al least one glomerulus with perihilar hyalinosis (as seen in this image), accompanied or not by sclerosis, and more than 50% of glomeruli with segmental lesions must have sclerosis and/or perihilar hyalinosis. There must not be glomeruli with collapsing or hypercellular lesions. (Mason’s trichrome, X400).
See Case 168 of our Case Series: Perihilar variant of FSGS
FSGS cellular variant
In order to diagnose this variant we must exclude the “tip” and the collapsing variants; in other words, if there is at least one glomerulus with tip lesion or collapsing lesion this variant is excluded.
It is defined by the presence of at least one glomerulus with endocapillary hypercellularity involving at least 25% of the tuft and causing occlusion of the capillary lumen/lumina. Any segment may be affected. The endocapillary cells include endothelial cells, macrophages and foam cells. Also lymphocytes and polymorphous can be identified. The cells occasionally manifest apoptosis, producing pyknotic or karyorrhectic debris. Endocapillary fibrin occasionally is identified, but without associated rupture of the glomerular basement membrane (in these cases we must suspecting a necrotizing glomerulonephritis). Podocyte hypertrophy and hyperplasia are typically identified overlying these lesions, but are not required features
Exclusion of cases wit tip lesion is based on the observation that in many cases this lesion is cellular.
In many texts the term “cellular lesion” is used to denominate hyperplasia of podocytes. This alteration must not be confused with the cellular variant in the present classification.
In my opinion, this variant is somewhat confusing, difficult to diagnose, and some authors do not record cases in their series. From a prognostic point of view, there appears to be no difference with the NOS variant. I only diagnose it in cases of complete nephrotic syndrome, undoubtedly negative immunofluorescence (or IgM and / or C3 in segmental lesions - nonspecific) and after an exhaustive analysis to rule out other glomerulopathies.

Figure 9. The arrows indicate a segment with increase of the cellularity and diminution or loss of the capillary lumina; the hypercellularity is due to proliferation of intrinsic glomerular cells and inflammatory cells that have migrated to the tuft, in this case mononuclear (lymphocytes and monocytes). Sometimes we can find polymorphous. In the case of the microphotography we found segmental and focal sclerosing lesions, NOS type, in 4 of 18 glomeruli, and only one (the one of the photo) with features of hypercellular variant. (H&E, X400)..
FSGS tip variant
The tip domain is the glomerular tuft zone adjacent to the proximal tubule: outer 25% of tuft next to origin of proximal tubule.
In order to diagnose this change we must exclude the collapsing variant, in other words, if there is at least one glomerulus with the characteristics collapsing lesion the tip variant is excluded. In addition, if there are lesions in perihilar segments the tip variant is excluded, having in consideration the peripheral nature of the lesions in the tip variant.
It is defined by the presence of at least 1 segmental lesion involving the tip domain with either adhesion between the tuft and Bowman’s capsule at the tubular lumen or neck, or confluence of podocytes with parietal or tubular epithelial cells at the tubular lumen or neck. The proximal tubular pole must be identified in the defining glomerulus. In some cases, the affected segment appears herniated into the tubular lumen. Segmental lesions may be characterized by endocapillary hypercellularity or sclerosis. Foam cells are common. Hyalinosis is variable. There often is podocyte hypertrophy/hyperplasia overlying the involved segment.
In other glomeruli there can be sclerosing lesions or hypercellularity in sites diverse to the tip domain, nevertheless, they must not compromise perihiliar segments.

Figure 10. In this glomerulus we can appreciate the characteristic location of the tip lesion. We can see adhesion to Bowman’s capsule and sclerosis. In other cases we can see hyaline segments or endocapillary hypercellularity in this portion of the tuft. (Masson trichrome, X400).

Figure 11. In this glomerulus the tip lesion is characterized by adhesion of the tuft to Bowman’s capsule near to the tubular neck. (Methenamine-silver, X400).
This variant has been associated, in different works, with a better prognosis (smaller risk of terminal renal failure) (Howie AJ, et al. Glomerular tip changes in childhood minimal change nephropathy. Pediatr Nephrol. 2008 Aug;23(8):1281-6. [PubMed link]). In our series of Hispanic patients we do not found a “favorable prognosis” for this variant and clinical outcome was almost similar to FSGS NOS (Arias LF, et al. Tip variant of focal segmental glomerulosclerosis: outcome and comparison to 'not otherwise specified' variant. Nephrol Dial Transplant. 2011;26(7):2215-21. [PubMed link]). It is my opinion that this variant should not be considered "benign".
(See Case 9 and Case 49 of our Case series)
FSGS collapsing variant (many of us consider this glomerulopathy as a glomerular disease other thanFSGS, not a variant of itt
This category excludes all the other variants. It is defined by at least 1 glomerulus with collapse and overlying podocyte hypertrophy and hyperplasia.
The capillary walls present retraction and
collapse. The lesion may be segmental or global and may involve peripheral or
perihilar segments. The number of affected glomeruli is very variable. Adhesions to the Bowman’s capsule and hyaline lesions are unusual. There may be other lesions in some glomeruli: sclerosis, hypercellularity, tip lesion
or global sclerosis.

Figure 12. See the glomerular tuft collapse, without conserved capillary lumina, with an irregular aspect and wrinkling of the capillary walls and with marked hypertrophy and hyperplasia of podocytes. This case corresponds to a 37-years-old male patient with NS, HIV negative, and without other predisponent factors: primary collapsing FSGS (idiopathic). (Methenamine-silver, X400).

Figure 13. Collapsing lesions can not be global and involve only some segments of the tuft. Although in this case the lesion is global, see greater collapse and podocyte hypertrophy and hyperplasia in the segments indicated with arrows (Silver, X.400).
This pattern has been associated with primary FSGS, HIV, toxicity by pamidronate, parvovirus B19, hemodynamic factors and other rare associations. (Albaqumi M, Barisoni L. Current Views on Collapsing Glomerulopathy. J Am Soc Nephrol. 2008;19(7):1276-81. [PubMed link]; Gulati A, et al. Idiopathic collapsing glomerulopathy in children. Clin Exp Nephrol. 2008;12(5):348-53. [PubMed link]).
In patients with HIV infection and this variant is frequent to find tubuloreticular inclusions in endothelial cells (electron microscopy) that are not identified in other collapsing lesions not associated to HIV; this feature is not exclusive of HIV-associate nephropathy and may be also identified in other diseases like lupus nephritis.
This variant seems to have a more aggressive course, with more severe proteinuria and faster evolution to terminal renal failure. There is predilection by Afro-American patients.
The term collapsing glomerulopathy (CG) is now more extensively used that "collapsing variant of FSGS". CG is not only a glomerular or podocyte disease, but all renal epithelial cells may be affected. The degree of tubulointerstitial damage varies from case to case and appears more pronounced in those forms where intrinsic epithelial cell damage, such as viral infection, is the related factor (Albaqumi M, Barisoni L. Current views on collapsing glomerulopathy. J Am Soc Nephrol. 2008;19(7):1276-81. [PubMed link]). Attempts to identify pathogenic mechanisms in CG have raised a number of hypotheses; however, the mechanisms through which these disorders lead to diffuse podocyte injury are poorly understood. Some investigators have focused their attention on the mitochondria. Mutations in genes encoding proteins involved in the mitochondrial respiratory chain may lead to CG in both mice and humans. Given the crucial role of oxygen availability in mitochondria metabolism, these data suggest that podocytes may be highly sensitive to hypoxic conditions. Recent evidence that hypoxia-inducible factor upregulation in podocytes leads to their proliferation and dedifferentiation in HIV associated nephropathy is striking. Injured podocytes show loss of some podocyte markers, induction of expression of some molecules and transdifferentiation to macrophage lineage (Canaud G, et al. Glomerular collapse associated with subtotal renal infarction in kidney transplant recipients with multiple renal arteries.Am J Kidney Dis. 2010 Mar;55(3):558-65. [PubMed link]).
The term “collapsing glomerulopathy” was used for the first time in 1986, and it was assumed to be a variant of FSGS. However, CG is frequently an aggressive disease, with massive proteinuria and rapidly progressive renal disease. The notoriously more aggressive outcome in CG that in the other variants of FSGS, and the phenotypic features of podocytes suggesting a different pathogenesis, have led some authors to propose that CG is not a variant of FSGS (Howie AJ. Problems with 'focal segmental glomerulosclerosis'. Pediatr Nephrol. 2011;26(8):1197-205. [PubMed link]).
See Case 6 - Case 40 - Case 86 - Case 159 (Collapsing glomerulopathies) of our case series.
Bibliography
Homepage - - - Tutorial Index